【ALD文献】东方理工大学孙学良院士&深圳大学张雷/宋中心Angew:基于W-O-Ru界面耦合的应变工程抑制晶格氧活化以实现稳定酸性水电解
- 2026-07-16 05:42:15
【ALD文献】东方理工大学孙学良院士&深圳大学张雷/宋中心Angew:基于W-O-Ru界面耦合的应变工程抑制晶格氧活化以实现稳定酸性水电解阅读合集:『ALD研究文章』『ALD综述文章』『ALD/MLD基础知识』『非ALD界面改性文章』『机器学习』『其他改性文章』 
【研究重点】 开发高活性和耐用的酸性析氧反应(OER)电催化剂仍然是质子交换膜水电解(PEMWE)的核心挑战。 【研究背景】 质子交换膜水电解(PEMWE)是实现绿色氢气生产的关键技术,但其阳极析氧反应(OER)在强酸性、高电位环境下动力学缓慢,且催化剂易降解。钌(Ru)基催化剂因其相对低廉的成本和固有的高OER活性,被视为替代昂贵铱(Ir)基催化剂的理想选择。然而,Ru基催化剂在酸性OER过程中,其表面晶格氧容易被活化(即发生晶格氧介导机制,LOM),导致Ru过度氧化和溶解,稳定性和寿命远不能满足实际应用需求。 为破解Ru基催化剂活性与稳定性难以兼得的难题,应变工程被认为是一种有效策略。然而,单一的拉伸或压缩应变往往只能优化其中一个方面:拉伸应变虽能稳定晶格氧,但会削弱中间体吸附而降低活性;压缩应变虽能优化吸附能,却无法阻止晶格氧的热力学不稳定性。因此,能否通过特定的界面结构设计,利用其在工况下的动态演变,实现在不同反应阶段对活性与稳定性的协同调控,成为突破这一瓶颈的关键方向。 基于此,研究团队提出了一种巧妙的“界面单元工程”策略。通过在RuO₂表面构建原子级分散且高度氧化的W₁O₃物种,形成强耦合的W-O-Ru界面单元。该设计的核心在于利用W原子独特的电子和几何结构,诱导RuO₂表面产生初始拉伸应变,同时通过电子耦合调控Ru的电子状态,并利用界面的适应性动态重构,以“同时”满足高活性和超高稳定性的苛刻要求。 【图文导读】 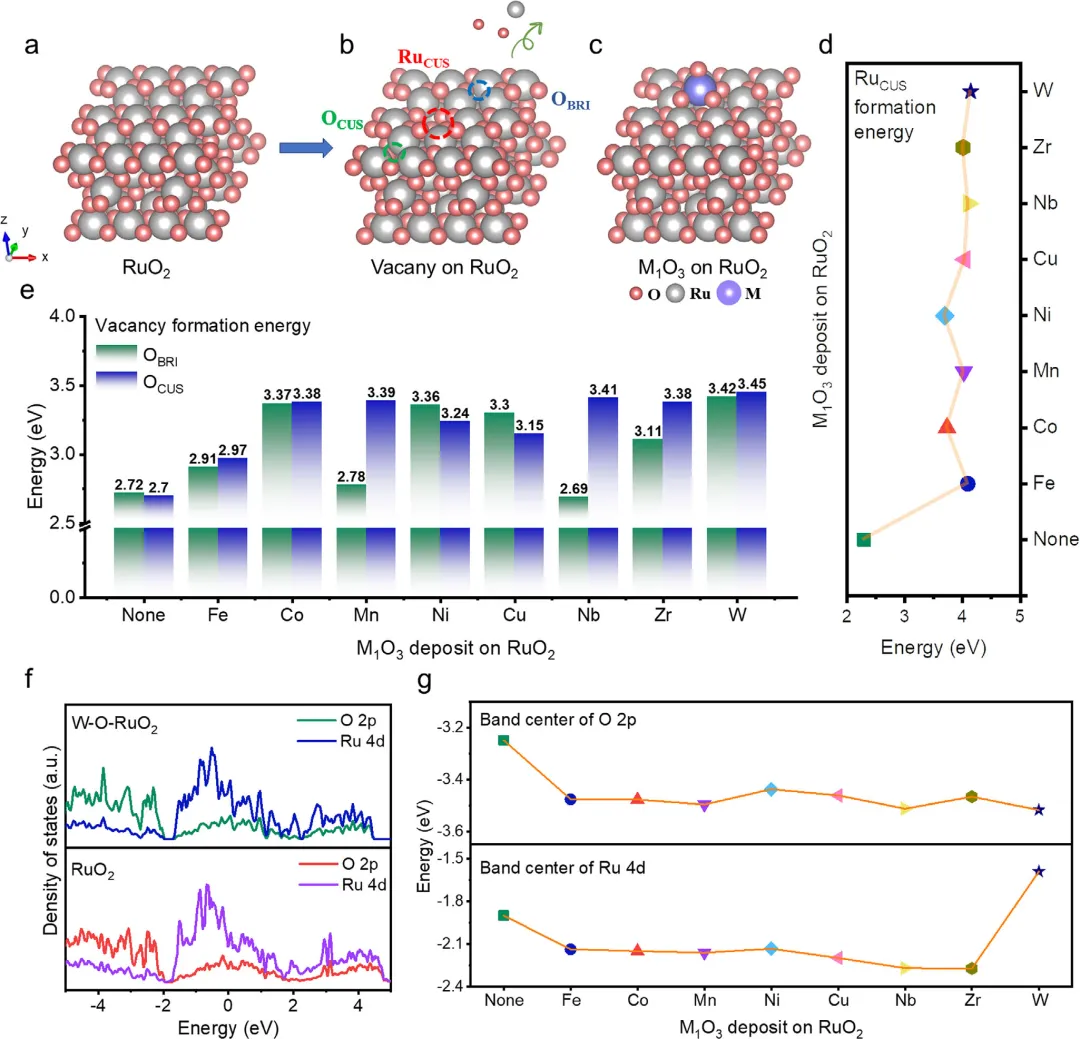
图1. 理论计算筛选与电子结构分析 图1通过密度泛函理论(DFT)计算,首先展示了纯RuO₂(110)表面易形成Ru空位和氧空位的原子结构。通过对八种不同过渡金属(M = Fe, Co, Mn, Ni, Cu, Nb, Zr, W)构建的W₁O₃修饰RuO₂模型进行系统性筛选,发现W-O-RuO₂拥有最高的RuCUS空位形成能(4.14 eV)和最高的O空位形成能(OBRI为3.42 eV,OCUS为3.45 eV),均远高于未修饰的RuO₂,证明了W₁O₃物种能最有效地抑制表面空位的形成。进一步通过态密度(PDOS)分析揭示,W-O-RuO₂具有最负的O 2p带中心(-3.52 eV),有效稳定了晶格氧;同时其Ru 4d带中心上移至-1.59 eV,预示了活性中心从氧向金属的转移,并且W 5d与Ru 4d的强耦合促进了电子离域,协同稳定了Ru活性中心。 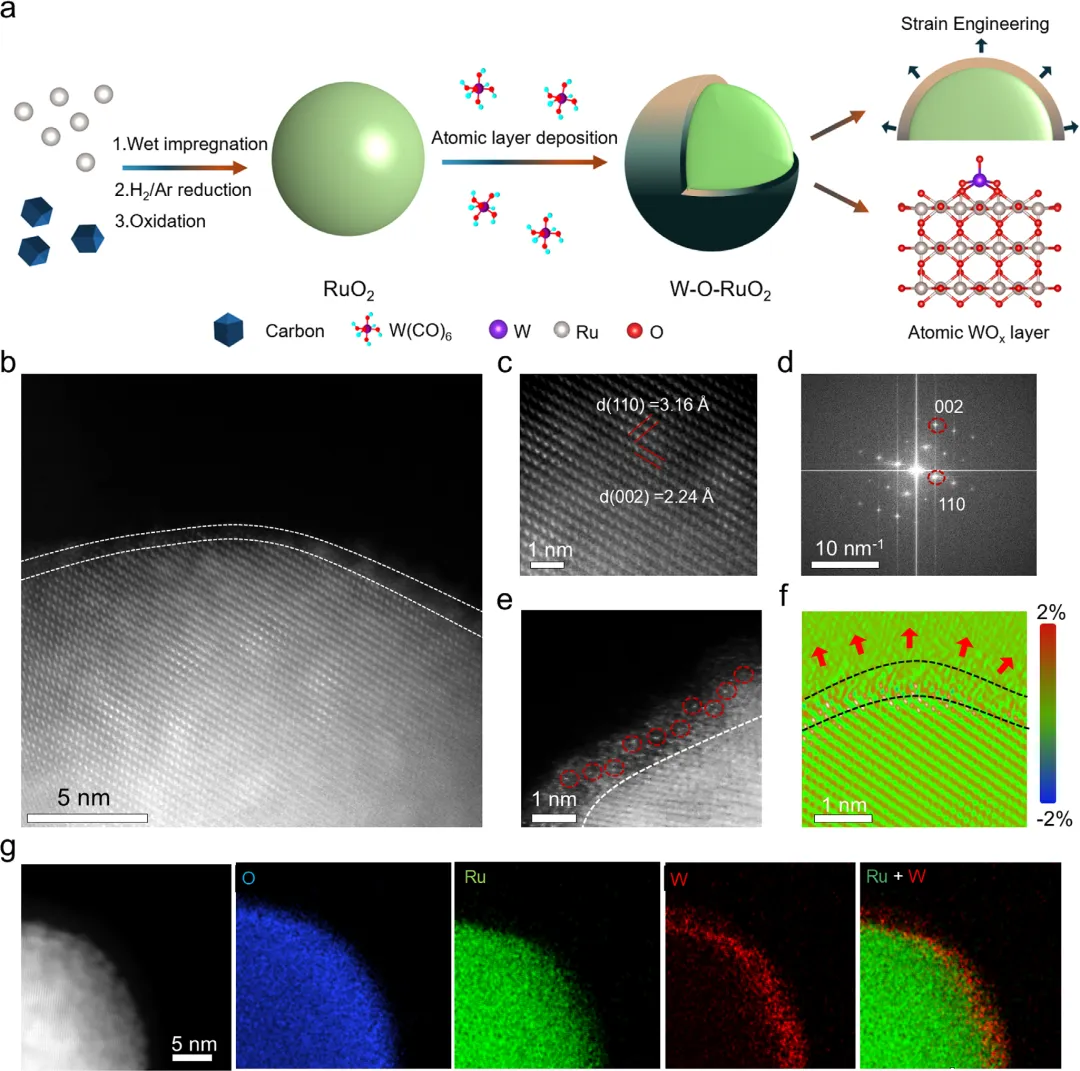
图2. W-O-RuO₂催化剂的合成与结构表征 图2通过示意图展示了从Ru/C纳米颗粒出发,经过氧化和原子层沉积(ALD)精确构建W-O-RuO₂催化剂的“自下而上”合成策略。高角度环形暗场扫描透射电镜(HAADF-STEM)图像清晰显示,在保持了完整的金红石RuO₂晶格(如(110)面和(002)面)之上,成功沉积了一层亚纳米级的非晶W-O覆盖层,形成了原子分散的W-O-Ru界面单元。关键的几何相位分析(GPA)量化了这种原子级界面耦合产生的局部表面拉伸应变(约2%),这源于W₁O₃配位环境与RuO₂基底的晶格失配。能谱(EDS)元素映射进一步证实W元素均匀且严格地分布在RuO₂表面,未进入体相晶格,证实了该界面耦合策略的普适性。 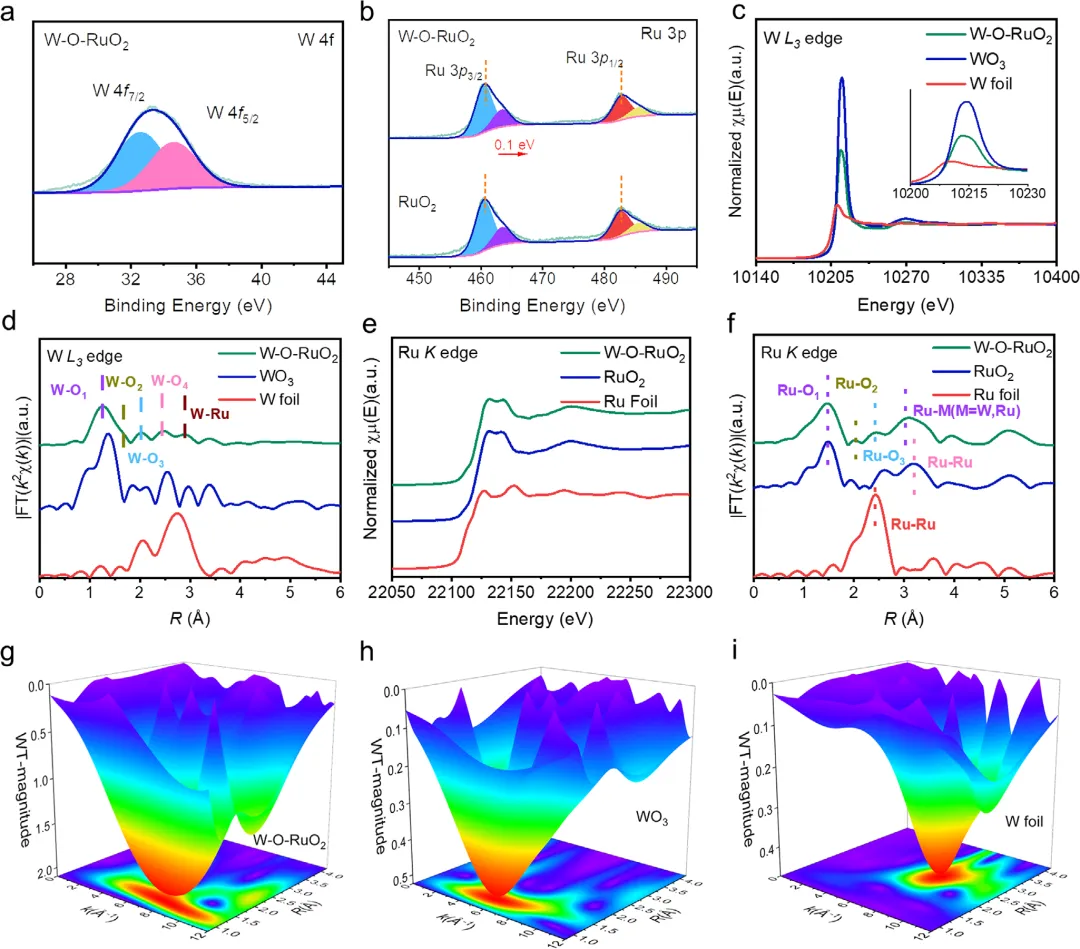
图3. W-O-RuO₂催化剂的电子结构与配位环境分析 图3结合X射线光电子能谱(XPS)和X射线吸收谱(XAS)系统揭示了催化剂的电子与几何结构。XPS显示W以W⁴⁺形式存在,且Ru 3p峰相比纯RuO₂向高结合能方向偏移0.1 eV,证实了W与Ru之间强烈的电子耦合。W L₃边X射线吸收近边结构(XANES)分析表明W的平均价态为+3.3,扩展X射线吸收精细结构(EXAFS)拟合出W-O₁、W-O₂、W-O₃三个短键和W-Ru键(2.88 Å),完美匹配了理论上预测的W₁O₃配位环境,排除了W团簇或体相WOₓ的形成。Ru K边XANES显示其氧化态为+3.4,且EXAFS中Ru-O₂键长变化及Ru-M(M=W, Ru)配位环境的调制,进一步证实了界面拉伸应变和W-Ru强相互作用的存在。小波变换(WT)图也提供了各元素配位环境差异的直观证据。 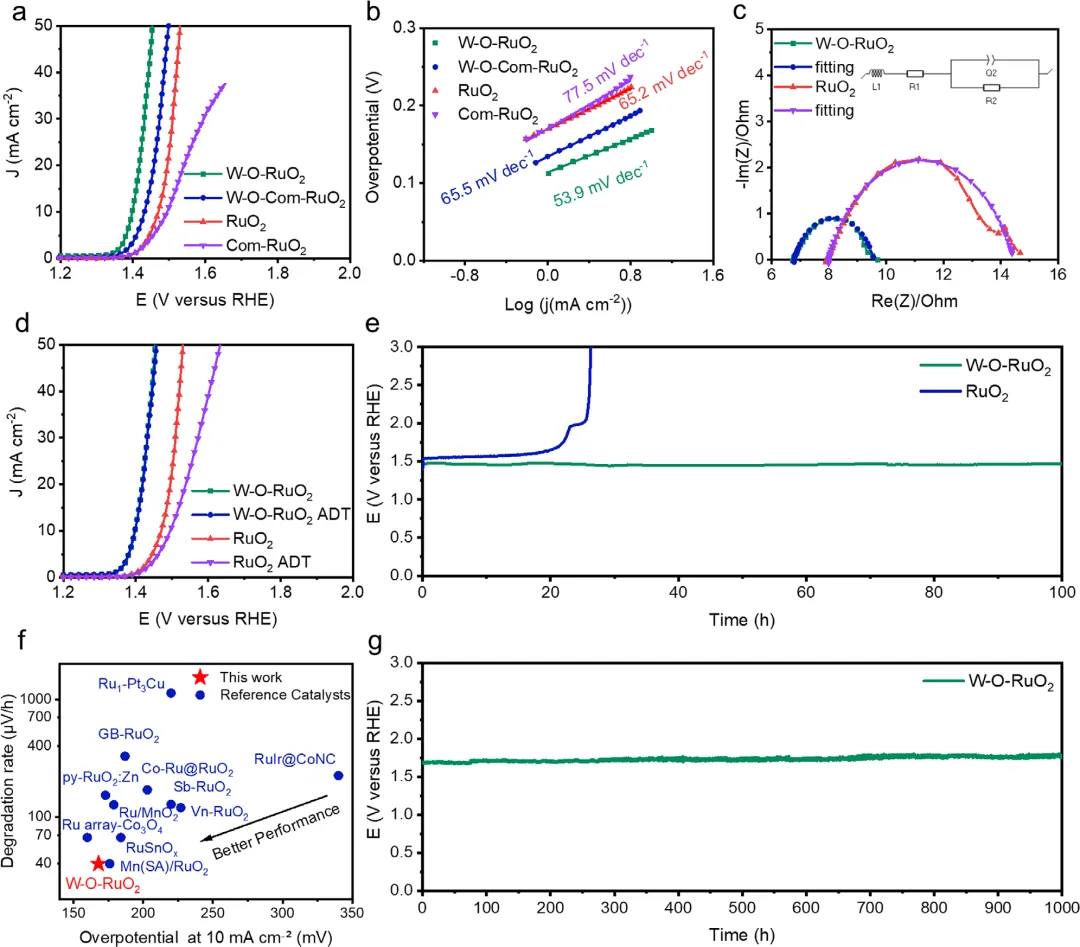
图4. W-O-RuO₂催化剂的电催化性能评估 图4全面评估了催化剂在酸性0.5 M H₂SO₄中的析氧反应(OER)性能。线性扫描伏安法(LSV)曲线表明,W-O-RuO₂仅需168 mV的过电位即可达到10 mA cm⁻²的电流密度,远优于纯RuO₂(240 mV),且具有最低的Tafel斜率(53.9 mV dec⁻¹)和电荷转移电阻(2.8 Ω),体现了优异的催化活性与动力学。加速耐久性测试(ADT)中,W-O-RuO₂在5000圈循环后极化曲线无衰减,在50 mA cm⁻²的恒电流(CP)测试中衰减率仅为40 μV/h,稳定性远超对照样。在质子交换膜水电解器(PEMWE)的实际应用中,使用W-O-RuO₂阳极仅需1.68 V即可实现1 A cm⁻²的工业级电流密度,并稳定运行超1000小时,衰减率低至63.3 µV/h,性能全面超越文献报道的先进Ru基催化剂。 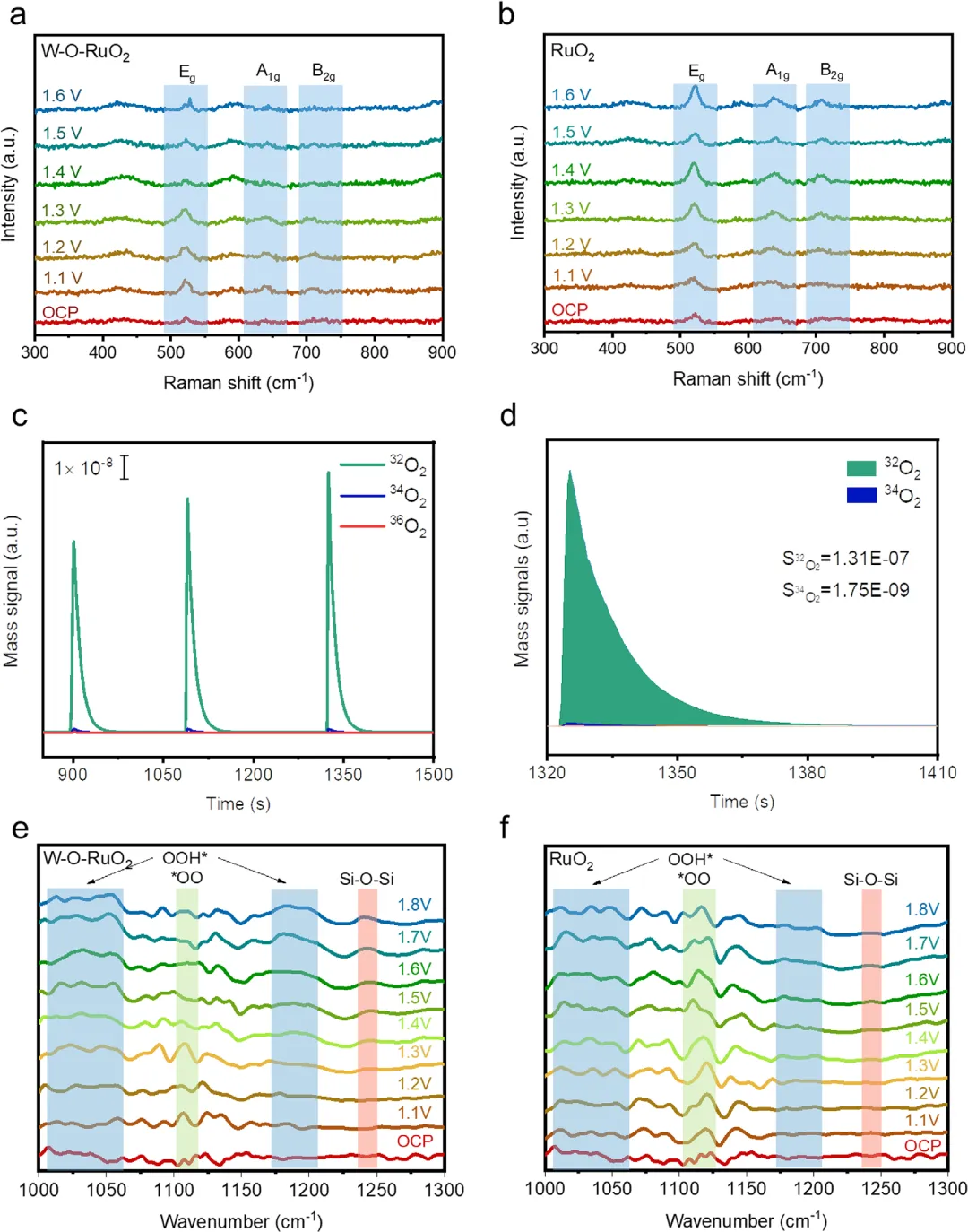
图5. W-O-RuO₂催化剂的反应路径机理分析 图5通过系列原位光谱技术揭示了W-O-RuO₂如何通过抑制晶格氧机制(LOM)实现稳定性。原位拉曼光谱显示,随着电位升高,W-O-RuO₂的金红石RuO₂特征振动峰(Eg, A₁g, B₂g)保持稳定,而纯RuO₂则出现明显红移和强度减弱,表明W-O界面单元有效抑制了晶格畸变和氧空位形成。原位差分电化学质谱(DEMS)结合¹⁸O同位素标记实验发现,W-O-RuO₂产生的³⁶O₂信号可忽略不计,经计算仅有0.65%的O₂来自晶格氧,远低于传统RuO₂。原位衰减全反射表面增强红外吸收光谱(ATRSEIRAS)进一步直接观察到,在W-O-RuO₂表面,代表吸附物析出机制(AEM)的OOH中间体信号(1033和1200 cm⁻¹)占主导,而代表LOM的OO信号(1122 cm⁻¹)很弱且在高电位消失。这些结果共同证实,原子层W覆盖层将反应路径从易降解的LOM成功切换至更稳定、可逆的AEM。 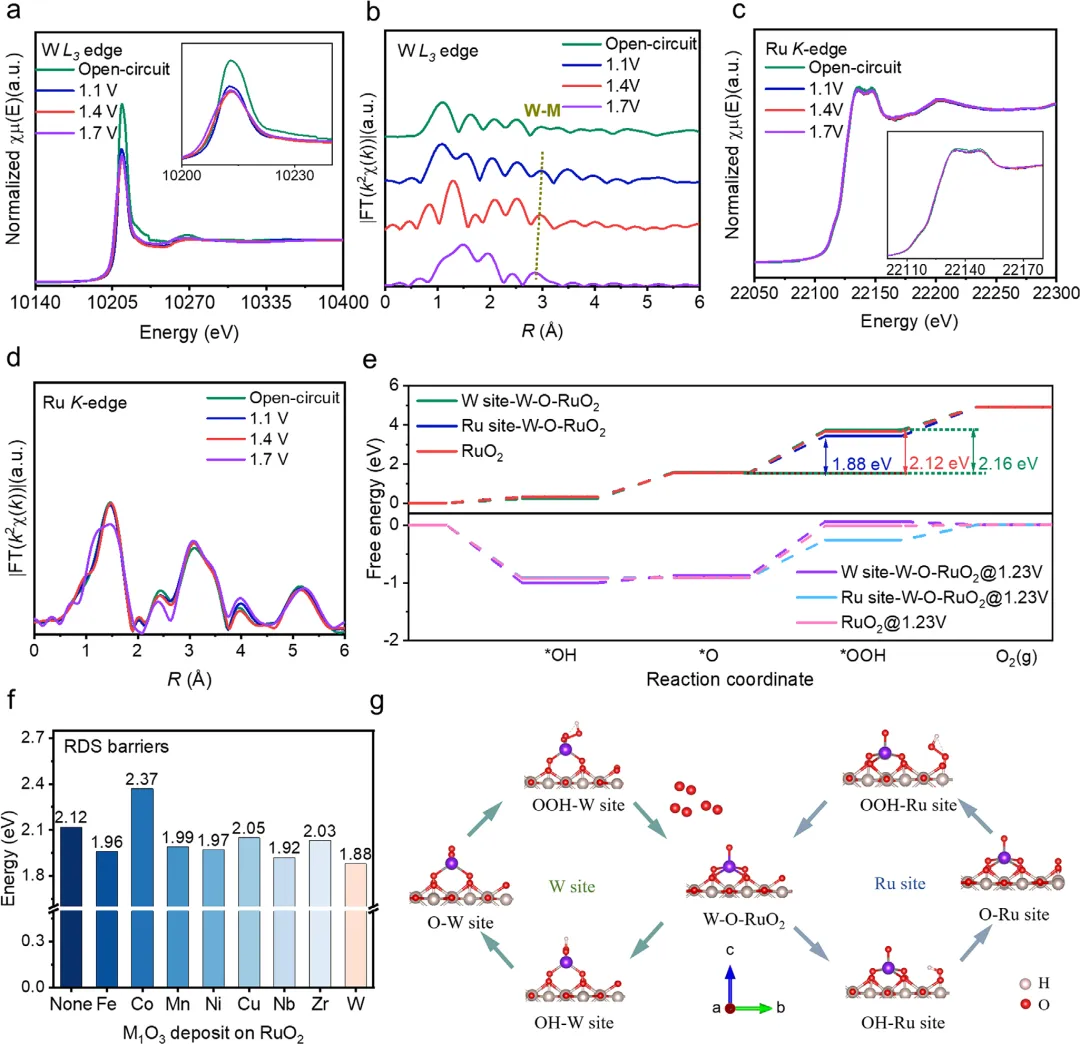
图6. W-O-RuO₂催化剂的增强活性机理分析 图6结合操作态XAS与DFT计算,揭示了催化活性增强的根源。操作态W L₃边EXAFS显示,随着施加电位从1.1 V升至1.7 V,W-Ru散射峰从~2.9 Å系统性地收缩至~2.8 Å,表明W-O-Ru界面在外电场作用下发生了动态的键长收缩,演化成一种压应变状态,这是界面强电子相互作用的直接证据。同时,Ru K边谱图无显著变化,证实了体相RuO₂的结构完整性。DFT自由能计算表明,W-O-RuO₂上的OER遵循一个精妙的W-Ru双位点协同机制:W位点主要负责初始的水活化和最终的产物脱附,而Ru位点在关键的O到OOH速率决定步骤(RDS)中展现出最低的能垒(1.88 eV),远低于W位点(2.16 eV)或纯RuO₂(2.12 eV)。这种协同作用是W通过界面键作为电子缓冲器来优化Ru电子结构的结果。 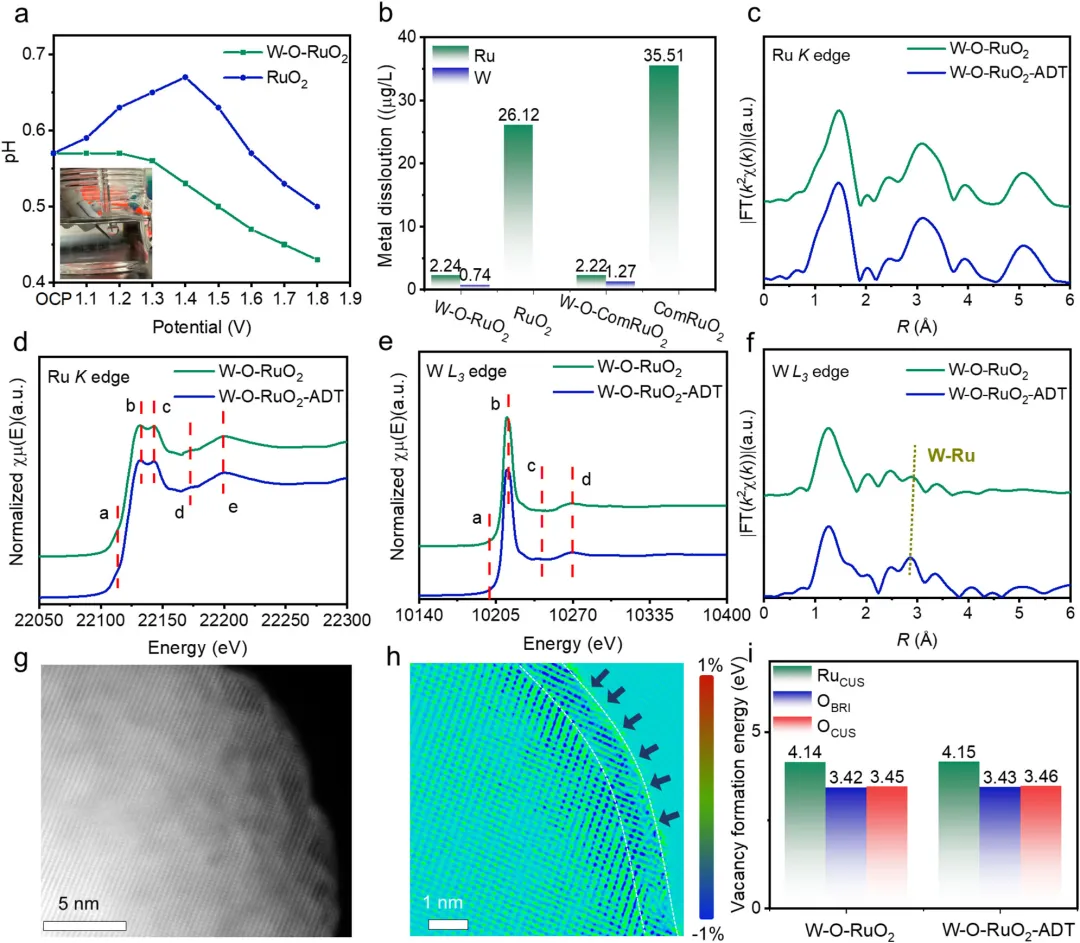
图7. W-O-RuO₂催化剂的稳定性机制与动态演变分析 图7系统阐述了W-O-RuO₂优异稳定性的根源。操作态pH测量和ICP-OES结果表明,W-O-RuO₂在OER过程中能有效抑制电解液酸化和Ru溶出(Ru溶出量仅为2.24 µg/L,远低于RuO₂的26.12 µg/L)。加速老化测试(ADT)后的XAS和TEM分析显示,W-O-RuO₂-ADT的Ru K边和W L₃边谱图与新鲜样品几乎完全一致,证实了其体相结构、W₁O₃配位环境和W的电子态均未发生退化。唯一的微妙变化是W-Ru散射峰进一步轻微收缩至~2.85 Å,对应的GPA分析也显示表面应变从初始的~2%拉伸应变转变为~1%的压缩应变。DFT计算表明,这种应力转变后的压缩态W-O-RuO₂-ADT拥有更高的空位形成能和更低的RDS能垒(1.81 eV)。结论是,原子工程化的W₁O₃覆盖层建立了一个强耦合的W-O-Ru界面,既能(i)抑制晶格氧活化,(ii)防止Ru溶解,(iii)维持结构完整性,又能(iv)动态适应界面应变,从而实现了卓越的长周期稳定性。 【总结】 本研究成功设计并构建了一种具有原子级W-O-Ru界面单元的W-O-RuO₂催化剂,有效解决了酸性OER中Ru基催化剂活性与稳定性难以兼得的难题。通过理论筛选与原子层沉积技术,实现了W₁O₃物种在RuO₂表面的精确构筑,该结构不仅通过强电子耦合使催化剂遵循更稳定的吸附物析出机制(AEM),同时动态抑制了晶格氧活化,还通过工况下的自适应性结构演化,使得催化界面从初始的拉伸应变转变为更稳定的轻微压缩状态。最终,该催化剂在酸性OER中展现出极低的过电位(168 mV @ 10 mA cm⁻²),并且在PEMWE中以1000 mA cm⁻²的电流密度稳定运行超过1000小时,实现了仅63.3 µV/h的超低衰减率,展现出巨大的实际应用潜力。 【启发/指导作用】


若您对原子层沉积设备有任何问题,欢迎扫码咨询!
【文献信息】 Yi Guan, X. Yao, R. Qi, et al. Angewandte Chemie International Edition (2026): e7876559. https://doi.org/10.1002/anie.7876559 DOI: 10.1002/anie.7876559
【免责声明】本文内容基于期刊上发表的高水平文章,旨在分享科学知识。更多详细信息,请查阅原文章。作者水平有限,如有错误,请联系删改。(如需快速获取文章的PDF文件,可后台联系管理人员) 欢迎关注,投稿/合作请联系:yhxu1031@163.com
在这里,结合理论指导设计、原子层工程和操作光谱来创建结构坚固、机械调谐的钌基催化剂。密度泛函理论表明,将 W1O3 沉积到 RuO2 上可以最大化 Ru 和 O 空位形成能,优于其他测试的过渡金属。 在此指导下,采用原子层沉积在 RuO2 (W-O-RuO2) 上构建原子耦合的 W-O-Ru 界面单元,产生拉应力表面,同时保留金红石核。 综合原位光谱和质谱分析表明,这种结构有效地抑制了晶格-氧活化,将反应从晶格-氧机制转变为更可逆的吸附质演化机制。Operando X 射线吸收光谱证实了 OER 过程中 W-O-Ru 界面的动态稳定性,该界面会演变成有弹性的轻度压缩 (1%) 状态而不会降解。 因此,W-O-RuO2 在 10 mA cm−2 下仅需要 168 mV 的过电势,并在 PEMWE 器件中维持 1 A cm−2 1000 小时,并且具有 63.3 μV/h 的超低降解率。 这项工作将界面单元工程建立为设计异常稳定的酸性 OER 催化剂的通用蓝图。
提出了“界面单元工程”策略,为催化剂设计提供了新范式:区别于传统的元素掺杂或构建核壳结构,该工作通过在活性材料表面精确定制一个原子尺度、结构明确的“界面单元”(如W₁O₃-RuO₂),实现了对电子结构、反应路径和应力状态的精准与协同调控。这启示研究者,未来的催化剂设计可以更加关注于构建这种具有特定功能的、原子级精确的表面/界面结构。 揭示了“动态应变适应”机制,为解耦活性-稳定性矛盾提供了新机理:该发现表明,催化剂在工况下并非静态的,其界面结构可以发生自适应演变(如从拉伸到轻微压缩)。这种动态响应能力使得催化剂能够“智能”地满足不同反应步骤对电子和几何结构的需求,从而突破传统静态应变工程中活性与稳定性的固有制衡。这鼓励研究者应更多地结合操作态表征手段,去发现和理解催化剂的“动态”本质。 强调了“反应路径调控”是实现稳定性的关键:该工作证明,通过界面工程,成功使催化剂不再依赖易导致结构退化的晶格氧介导机制(LOM),而是转向更可逆、更稳定的吸附物析出机制(AEM)。这为开发高稳定性催化剂提供了一个明确的指导方向:未来的催化剂设计应将“抑制LOM、促进AEM”作为一项核心目标,而原位光谱和质谱技术是验证该目标的必需工具。 展示了从“理论预测”到“精准合成”再到“机理验证”的闭环研究方法:从DFT高通量筛选,到ALD精准沉积,再到多维度原位/操作态谱学表征,本研究提供了一个高度完整和严谨的科研范式。这启示研究者,未来复杂催化体系的开发,必须走通这种集成的、多学科交叉的研究路径,才能系统性地、有根据地攻克核心科学难题,而非进行盲目的试错。
相关设备推荐:
还在为薄膜沉积 “精度不足、3D 高深宽比结构难适配、设备笨重难集成” 发愁?Quantum Design中国引进的GEMStar XT 台式三维原子层沉积系统重磅破局!以 82×64×31cm 小巧机身集成全功能,攻克传统设备温度不均、沉积受限等痛点,支持 1500:1 高深宽比 3D 结构与多尺寸基片均匀沉积,凭 99.9% 温度均匀性、超 99% 膜厚均匀性及 ALD/MLD 兼容能力,为科研提供原子级精准薄膜解决方案!
核心优势:
ALD+MLD双模一体:标配稳定ALD无机高精度沉积,支持MLD有机沉积升级,可构筑有机/无机复合界面,适配锂电等高端界面改性研究;
8 路前驱体通道适配复杂 / 掺杂薄膜;
多片同步沉积提效,侧面手套箱集成不占地、无热干扰;
超高沉积精度:埃级-纳米级精准可控,可按需定制薄膜厚度、成分、结构,支持单分子层至多层复合薄膜构筑;
等离子增强型可沉积难成膜材料,操作便捷且数据可溯源。
应用领域:
覆盖量子点器件、钙钛矿太阳能电池、锂电池、催化材料、半导体领域等,精准获取薄膜成分均匀性、结构稳定性等核心数据,助力高水平成果产出,是纳米技术、光电材料研究的硬核工具!
本文来自网友投稿或网络内容,如有侵犯您的权益请联系我们删除,联系邮箱:wyl860211@qq.com 。
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 深圳市十五五规划:深化创业板改革,科创债、专项债双线发力
- 深圳200w刚需买房:康达尔花园“云”看房笔记(龙岗大芬地铁站)
- 深圳光明文化艺术中心剪影
- 深圳中考生,加油!
- 藏在深圳的免费古寺生态园|地铁直达!古风出片+玩水捞鱼+挖沙露营+儿童乐园+祈福
- 【产业政策】深圳市科技创新局关于印发《深圳市具身智能机器人技术创新与产业发展行动计划(2025-2027年)(修订版)》的通知
- 深圳重磅新规!逾期被爆通讯录、P 图羞辱,催收直接定性黑恶势力,负债人维权全流程)
- 深圳工业总会一行走访旷真律所,共探法律赋能企业新路径
- 【深圳招聘】深圳政法委下属事业单位招聘5名员额制人员!应往届均有岗位,7月1日截止,报名从速~
- 深圳街头 l 民治: 城中村的日常
