深圳大学骆静利院士、大湾区大学奚修安研究员等ACB丨FeO₆八面体调控与自旋态协同解锁La₀.₅Sr₀.₅FeO₃中CO₂电解动力学

文章标题:Synergistic FeO₆ Octahedral Regulation and Spin-State Tuning in La₀.₅Sr₀.₅FeO₃ for Enhanced CO₂ Electrolysis Kinetics in SOECs(La₀.₅Sr₀.₅FeO₃中FeO₆八面体调控与自旋态协同增强SOEC中CO₂电解动力学)
第一作者:Jie Gao
通讯作者:Xiuan Xi(奚修安), Jing-Li Luo(骆静利)
通讯作者单位:
- 深圳大学土木与交通工程学院
- 深圳大学材料科学与工程学院,深圳市能源电催化材料重点实验室
- 大湾区大学物理科学学院、工程学院
论文DOI:10.1016/j.apcatb.2026.126780
期刊:Applied Catalysis B: Environment and Energy, 2026
研究背景
固体氧化物电解池(SOEC)可在高温下将CO₂电化学还原为CO,是实现碳循环和可再生能源存储的关键技术。然而,传统Ni基阴极虽活性高,但易因积碳失活;钙钛矿氧化物(如LaFeO₃)因具有优异的抗积碳性、混合离子-电子导电性和结构可调性而备受关注,但其对惰性CO₂分子的吸附活化能力有限,本征活性不足。
近年来研究表明,钙钛矿中BO₆八面体的几何构型(键长、键角、畸变程度)直接影响B位过渡金属3d轨道与O 2p轨道的重叠,进而调控金属-氧共价性和氧离子迁移率。同时,B位过渡金属的自旋态(高自旋HS、低自旋LS)也决定了反应中间体的吸附能和电子转移效率。然而,大多数研究仅关注其中单一因素,八面体几何与自旋态的协同效应对CO₂电解动力学的影响仍未被系统探索。
本文通过在LaFeO₃的A位进行Sr²⁺部分取代La³⁺,同时调控FeO₆八面体几何和Fe的自旋态。Sr掺杂使Fe-O-Fe键角从158.2°增至176.2°,键长从2.002 Å缩短至1.956 Å,强化了Fe 3d-O 2p杂化和Fe-O共价性;同时,电荷补偿诱导Fe³⁺氧化为Fe⁴⁺,增加了高自旋Fe⁴⁺(t₂g⁴eg¹)和五配位HS Fe³⁺的比例,优化了eg轨道占据。最优组成La₀.₅Sr₀.₅FeO₃(LSFO)在800°C、1.5 V下电流密度达2.37 A·cm⁻²,是未掺杂LaFeO₃(LFO)的2倍,且稳定运行超200小时。
全文速览
本文系统研究了Sr掺杂量(x=0, 0.25, 0.5, 0.75, 1)对La₁₋ₓSrₓFeO₃结构、电子构型和CO₂电解性能的影响。XRD Rietveld精修表明,随着Sr含量增加,材料从正交(LFO)→立方(LSFO)→四方(SFO)转变。LSFO(x=0.5)为立方相,Fe-O-Fe键角接近180°,Fe-O键长最短。
电化学测试显示,LSFO阴极在800°C、1.5 V下电流密度2.37 A·cm⁻²,极化电阻(Rp)仅0.174 Ω·cm²(@1.3 V),远低于LFO(0.401 Ω·cm²)。ECR测试表明LSFO的化学表面交换系数(Kchem=6.84×10⁻³ cm·s⁻¹)和化学扩散系数(Dchem=1.16×10⁻³ cm·s⁻¹)均显著高于LFO。XPS、EPR和TG证实LSFO具有更高氧空位浓度;CO₂-TPD显示其CO₂吸附能力更强。
XANES和EXAFS证实LSFO中Fe-O键长缩短、Fe价态升高。⁵⁷Fe Mössbauer谱揭示了HS Fe⁴⁺和五配位HS Fe³⁺的存在。DFT计算表明,LSFO的氧空位形成能(1.28 eV)远低于LFO(3.77 eV),CO₂吸附自由能(-0.34 eV)也更负。
核心发现:Sr掺杂通过缩短Fe-O键、增大Fe-O-Fe键角,强化Fe 3d-O 2p杂化,增强Fe-O共价性,降低氧空位形成能;同时,高价HS Fe⁴⁺提供了空eg轨道,通过双交换机制(Fe³⁺-O-Fe⁴⁺)促进电子快速迁移,而五配位HS Fe³⁺提供了不饱和配位活性位点,增强了CO₂吸附活化。几何调控与自旋态调控的协同作用共同解锁了LSFO的高本征活性。
文章亮点
⭐首次揭示几何-自旋协同机制:通过Sr掺杂同时调控FeO₆八面体几何和Fe自旋态,阐明了两者对CO₂电解动力学的协同增强作用。⭐精准的结构-活性关联:Rietveld精修、XANES/EXAFS、Mössbauer谱联合DFT计算,从原子尺度建立了“八面体畸变→Fe-O共价性→氧空位形成能→CO₂吸附/活化”的完整链条。⭐优异的电化学性能:LSFO在800°C、1.5 V下电流密度2.37 A·cm⁻²,是LFO的2倍;极化电阻降低56%;稳定运行超200小时。⭐多维度机理验证:ECR(表面交换/体相扩散)、DRT(弛豫时间分布)、EPR、CO₂-TPD、XPS、XAS、Mössbauer谱等多技术联用,证据充分。⭐理论计算与实验高度一致:DFT计算的氧空位形成能、CO₂吸附能与实验观察的氧空位浓度、CO₂-TPD结果完美匹配。
图文解析
图1. 结构表征
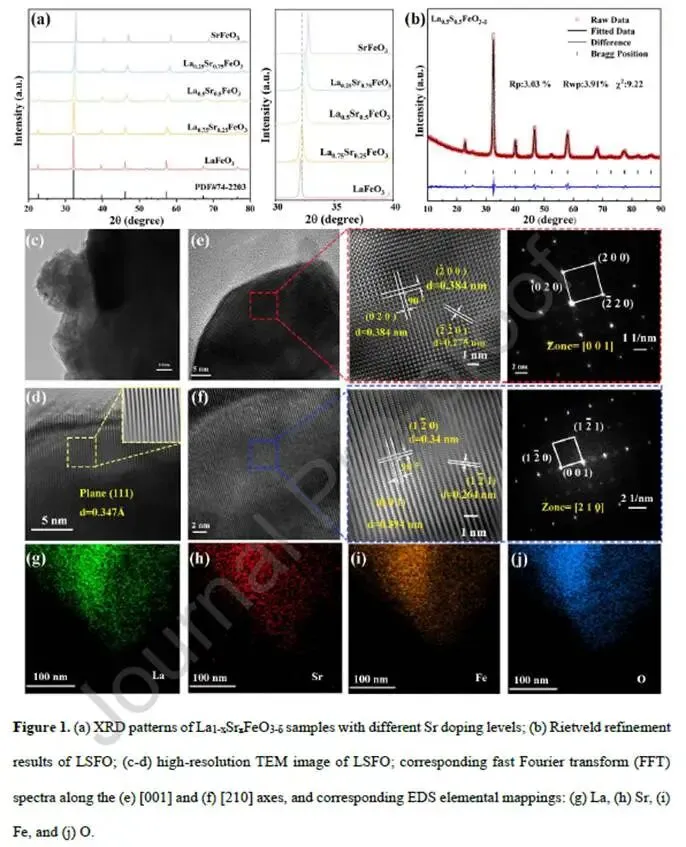
图1a XRD图谱:随Sr掺杂量增加,主衍射峰逐渐向高角度移动,表明晶格收缩。尽管Sr²⁺离子半径(1.44 Å)略大于La³⁺(1.36 Å),但电荷补偿诱导的Fe³⁺→Fe⁴⁺氧化(Fe⁴⁺半径0.585 Å < Fe³⁺半径0.645 Å)导致总体积减小。
图1b LSFO的Rietveld精修:LSFO为立方相(Pm-3m),而LFO为正交相(图S1),表明Sr掺杂显著提高了晶体对称性。
图1c-f HRTEM和FFT:LSFO的(111)晶面间距0.347 nm,FFT沿[001]和[210]轴证实其立方结构和高结晶度。
图1g-j EDS元素mapping:La、Sr、Fe、O均匀分布,无元素偏析。
解析:Sr掺杂诱导的结构演变(正交→立方)为后续FeO₆八面体几何调控奠定了基础。立方相中Fe-O-Fe键角接近180°,有利于d-p轨道“头对头”重叠。
图2. 电化学性能与阻抗分析
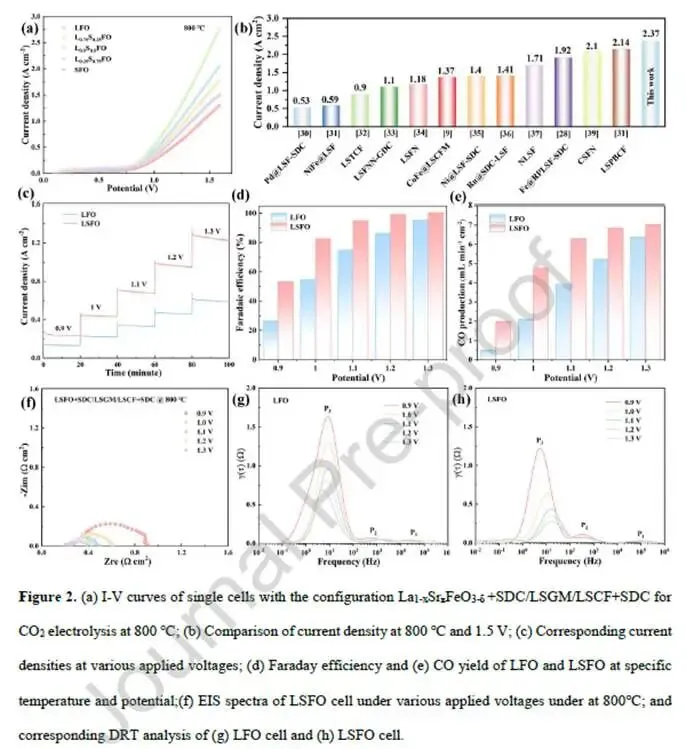
图2a I-V曲线(800°C,纯CO₂):随着Sr掺杂量增加,电流密度显著增大。x=0.5(LSFO)性能最优,x=0.75和1时略有下降,呈“火山型”趋势。
图2b对比LSFO与LFO:LSFO在1.5 V下电流密度2.37 A·cm⁻²,LFO仅1.14 A·cm⁻²,提升约2倍。
图2c稳定性测试(200小时,1.2 V):LSFO电流密度稳定在约0.85 A·cm⁻²,无衰减,而LFO持续下降。SEM/EDS(图S8)显示LSFO+SDC电极与LSGM电解质界面结合良好,无分层或开裂。
图2d-e FE和CO产率:随电流密度增加,FE维持在90%以上,CO产率线性上升,表明CO₂还原为CO的高选择性。
图2f EIS(0.9-1.3 V):LSFO和LFO的欧姆电阻相近(~0.3 Ω·cm²),但LSFO的极化电阻(Rp)从0.648降至0.174 Ω·cm²,远低于LFO(1.08→0.401 Ω·cm²)。
图2g-h DRT分析:三个峰分别对应离子传导(P₁,高频)、电荷转移(P₂,中频)、电极极化/气体吸附扩散(P₃,低频)。P₃为决速步,LSFO的P₃峰强度显著低于LFO,且随电压增加快速减弱,表明Sr掺杂加速了表面反应动力学。
解析:LSFO在活性、稳定性和动力学上均显著优于LFO。DRT精准定位了性能提升的关键在于低频区的电极极化过程(CO₂吸附、活化、CO脱附)。
图3. 表面化学与氧空位
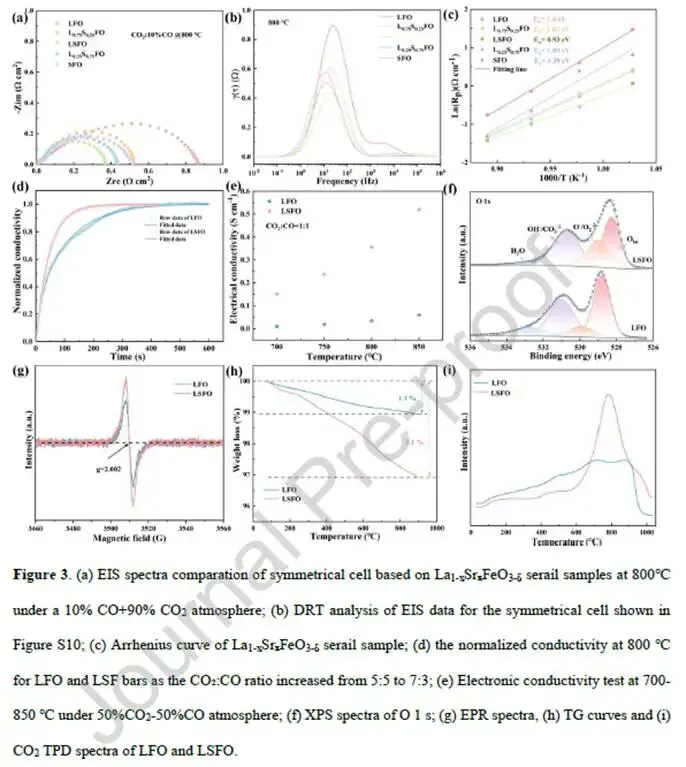
图3a对称电池EIS(800°C):LSFO的Rp=0.36 Ω·cm²,比LFO(0.87 Ω·cm²)降低56%。
图3b DRT分析(对称电池):随Sr掺杂量增加,低频峰强度先降后升,x=0.5时最低,与I-V趋势一致。
图3c Arrhenius图:LSFO的活化能E_a=0.93 eV,低于LFO(1.40 eV),表明反应能垒降低。
图3d ECR(归一化电导率弛豫):当CO₂:CO从5:5升至7:3时,LSFO的归一化电导率更快达到平衡。拟合得LSFO的Kchem=6.84×10⁻³ cm·s⁻¹,Dchem=1.16×10⁻³ cm·s⁻¹;LFO仅5.74×10⁻³和1.83×10⁻⁴ cm·s⁻¹。Kchem提升19%,Dchem提升6.3倍。
图3e电子电导率(CO-CO₂气氛,700-850°C):LSFO电导率0.34 S·cm⁻¹(800°C),LFO仅0.035 S·cm⁻¹,提升约10倍。LSFO的活化能0.86 eV低于LFO的1.19 eV。
图3f XPS O 1s:LSFO的晶格氧峰(528.5 eV)相对于LFO(528.8 eV)负移,表明氧电子云密度增加,有利于氧空位形成。表面氧物种比例从59.88%增至69.03%。
图3g EPR(g=2.002):LSFO信号强度显著高于LFO,表明更高浓度的氧空位/未配对电子。
图3h TG:LSFO失重3.1%,LFO仅1.1%,进一步证实LSFO中氧空位更多。
图3i CO₂-TPD:LSFO在400-1000°C范围内CO₂脱附峰面积远大于LFO,表明更强的CO₂化学吸附能力。
解析:Sr掺杂大幅提升了LSFO的氧空位浓度、表面交换动力学、体相扩散和电子电导率,这些因素共同促进了CO₂的吸附和活化。
图4. DFT计算:八面体几何与CO₂活化
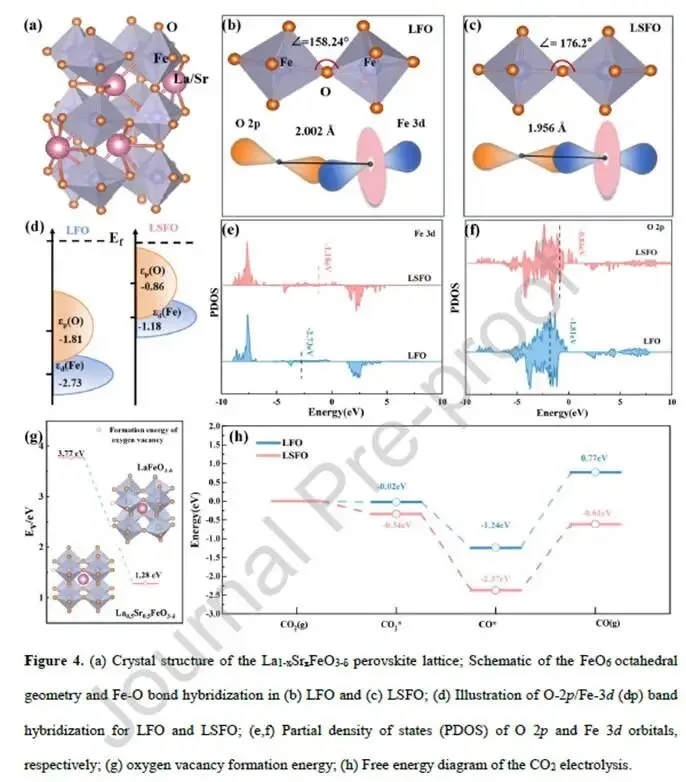
图4a La₁₋ₓSrₓFeO₃晶格结构示意图。
图4b-c FeO₆八面体几何对比:
- LFO(正交):Fe-O-Fe键角158.24°,Fe-O键长2.002 Å,八面体严重倾斜。
- LSFO(立方):Fe-O-Fe键角176.2°,Fe-O键长1.956 Å,接近理想“头对头”构型,Fe-O共价性增强。
图4d d-p带中心示意图:LSFO的d带中心(-1.18 eV)和p带中心相比LFO(-2.17 eV)分别上移1.55和0.95 eV,更接近费米能级,强化了d-p杂化。
图4e-f PDOS:LSFO中Fe 3d和O 2p在费米能级附近的态密度显著高于LFO,表明更高的电子可及性和反应活性。
图4g氧空位形成能(Fe-Vo-Fe位点):
- LFO:3.77 eV
- LSFO:1.28 eV(降低66%) → Sr掺杂极大促进了氧空位的形成,有利于氧离子迁移。
图4h CO₂电解自由能图:
- CO₂吸附:LFO ΔG=-0.02 eV,LSFO ΔG=-0.34 eV(更有利)
- 碳酸盐解离:LFO ΔG=-1.22 eV,LSFO ΔG=-2.03 eV(更有利) → LSFO在CO₂吸附和解离步骤均具有热力学优势。
解析:DFT从原子层面揭示了Sr掺杂的“双重红利”——几何上缩短Fe-O键、拉直Fe-O-Fe键角,增强共价性、降低氧空位形成能;电子上上移d-p带中心、提高态密度,增强CO₂吸附和活化。
图5. 电子结构:Fe价态、配位与自旋态
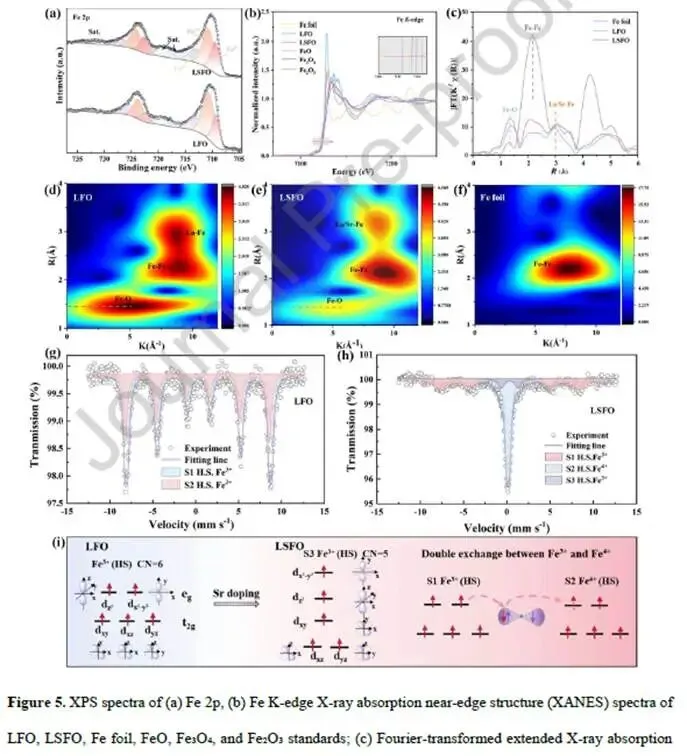
图5a Fe 2p XPS:LSFO中Fe²⁺/Fe³⁺/Fe⁴⁺共存,平均价态从LFO的2.75升至LSFO的2.95,与XRD峰位移一致。
图5b Fe K-edge XANES:LSFO的吸收边相对于LFO向高能移动(插图),证实Fe价态升高。
图5c FT-EXAFS:LSFO的Fe-O键长(1.86 Å)短于LFO(1.97 Å),与DFT和Rietveld结果一致。Fe-Fe配位峰也向短距离移动。
图5d-f小波变换(WT)EXAFS:LSFO的Fe-O散射强度更高,Fe-Fe散射峰更集中,证实Fe-O共价性增强和局部结构有序度提高。
图5g-h ⁵⁷Fe Mössbauer谱:
- LFO:两个六线峰(sextet),对应HS Fe³⁺(磁有序正交相)。
- LSFO:一个六线峰(S1,HS Fe³⁺,H=50.0 T)+两个双峰(doublet)。S2(IS=0.13 mm/s)归属HS Fe⁴⁺;S3(IS=0.32 mm/s)归属五配位HS Fe³⁺(与氧空位相邻)。 → 首次直接观测到Sr掺杂诱导的HS Fe⁴⁺和不饱和配位Fe³⁺。
图5i电子构型示意图:
- 左:LFO中HS Fe³⁺(t₂g³eg²),eg轨道占据2。
- 中:LSFO中HS Fe⁴⁺(t₂g⁴eg¹),eg轨道占据1,提供空轨道;同时通过双交换机制(Fe³⁺-O-Fe⁴⁺)实现eg电子快速迁移。
- 右:五配位HS Fe³⁺(因氧空位导致配位数降低),dx²-y²轨道能级升高,成为强Lewis酸位点,与CO₂的π*轨道产生强相互作用。
解析:Mössbauer谱是本工作的点睛之笔。它直接证明了Sr掺杂后Fe⁴⁺和五配位Fe³⁺的出现——前者通过双交换机制增强导电性,后者提供不饱和活性位点增强CO₂吸附。eg轨道占据从2优化至1,符合Shao-Horn等提出的“eg≈1”最优活性描述符。
总结与展望
本研究系统揭示了A位Sr掺杂对LaFeO₃基钙钛矿CO₂电解性能的协同增强机制,可总结为两条主线:
几何主线:Sr掺杂 → 晶格收缩 → 正交→立方相变 → Fe-O-Fe键角增大(158.2°→176.2°)→ Fe-O键长缩短(2.002→1.956 Å)→ Fe 3d-O 2p轨道“头对头”重叠增强 → Fe-O共价性增强 → 氧空位形成能降低(3.77→1.28 eV)→ 氧离子迁移率提升(Dchem提高6.3倍)。
自旋主线:Sr²⁺取代La³⁺ → 电荷补偿 → Fe³⁺氧化为Fe⁴⁺ → HS Fe⁴⁺(t₂g⁴eg¹)出现 → eg轨道占据从2优化至1 → 双交换网络(Fe³⁺-O-Fe⁴⁺)建立 → 电子电导率提升10倍;同时,氧空位诱导五配位HS Fe³⁺形成 → 不饱和配位点作为强Lewis酸中心 → CO₂吸附增强(ΔG从-0.02→-0.34 eV)→ CO₂解离能垒降低(ΔG从-1.22→-2.03 eV)。
两条主线在LSFO中协同作用,使800°C、1.5 V下的电流密度达到2.37 A·cm⁻²,是未掺杂LFO的2倍,且稳定运行超200小时。
未来展望:
1.将该“几何-自旋协同”策略拓展至其他钙钛矿体系(如LaCoO₃、LaNiO₃)和电催化反应(如OER、ORR)。
2.利用原位/operando XAS和Mössbauer谱,实时追踪CO₂电解过程中Fe自旋态和八面体几何的动态演化。
3.通过机器学习筛选A/B位掺杂组合,高通量预测最优eg占据和八面体畸变程度。
4.在更大尺寸单电池和电堆中评估LSFO阴极的长期稳定性,并探索其在实际工业CO₂尾气处理中的应用。
通讯作者简介
奚修安,大湾区大学物质科学学院研究员,日本大阪大学客座研究员、独立课题组组长。博士毕业于日本大阪大学,主要研究方向为固体氧化物燃料电池/电解电池等能源材料与器件的技术开发、应用及其机理研究,在固体氧化物燃料电池/电解电池的制备、封装、表征、性能测试及机理研究方面积累了10余年的研究经验,目前已在eScience, Nat. Commun., Angew. Chem. Int. Ed., Adv. Energy Mater., Adv. Func. Mater., Appl. Catal. B-Environ. Energy, Nano Energy, ACS Catal., J. Mater. Chem. A, J. Power Sources等国际知名期刊发表SCI论文60余篇,申请国家发明专利10余项。曾获得“国家优秀自费留学生奖学金”、“深圳市海外高层次人才”、“深圳市高层次专业人才”等荣誉称号。近年来主持或参与过国家自然科学基金-外国资深学者团队项目(试点)、国家自然科学基金面上项目、广东省自然科学金面上项目,深圳市孔雀团队项目、中国博士后科学基金特别资助及中国博士后科学基金面上项目一等资助等10余项科研项目。
骆静利(Jing-Li Luo):深圳大学材料学院全职特聘教授,博士生导师,加拿大国家工程院院士,中国腐蚀与防护学会外籍会士。曾任加拿大阿尔伯塔大学化学与材料工程系教授及加拿大替代燃料电池首席科学家。目前担任Electrochemical Energy Reviews (Springer Nature)、Corrosion Science和Corrosion Communications 编委,国际腐蚀理事会理事等职。她长期从事的研究领域包括固体氧化物燃料电池(SOFC)/电解池(SOEC)、碳基等能源分子转化电催化材料、腐蚀与防护等。发展了多类高效、高选择性、高稳定的能源分子转化,尤其是碳基分子转化电催化材料,开发建立了多种新型的SOFC/SOEC体系,在CO2和烷烃等碳基分子清洁转化利用等方面取得了多项突破性研究成果。已在国际顶尖期刊J. Am. Chem. Soc.、Nat. Commun.、Sci. Adv.、Adv. Mater.、Angew. Chem. Int. Ed.、Matter、Energy Environ. Sci. 等国际顶尖期刊发表论文470多篇,入选2022, 2023, 2024全球前2%顶尖科学家终身科学影响力榜单,且入围爱思唯尔2023年度“中国高被引学者”榜单。
声明:
文章内容仅代表本人观点,本文数据来源于Gao et al. Applied Catalysis B: Environment and Energy, 2026, DOI: 10.1016/j.apcatb.2026.126780论文,图片版权归原作者和出版社所有。如有侵权,请联系后台,感谢支持,欢迎批评指正!
欢迎各位老师同学们在公众号上投稿分享课题组研究成果,不限期刊和发表时间!后台联系即可。
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
