清华大学深圳国际研究生院余旷2025年人工智能驱动材料设计领域科研成果汇总
- 2026-06-17 10:14:23
1 长程分离型水分子力场的精细化与性能基准评估

英文标题:Refinement and Performance Benchmark for Range-Separated Water Force Field
作者:Qian Gao , Junmin Chen ∗ , and Kuang Yu †
原文链接:https://doi.org/10.48550/arXiv.2601.18416
DOI号:10.48550/arXiv.2601.18416
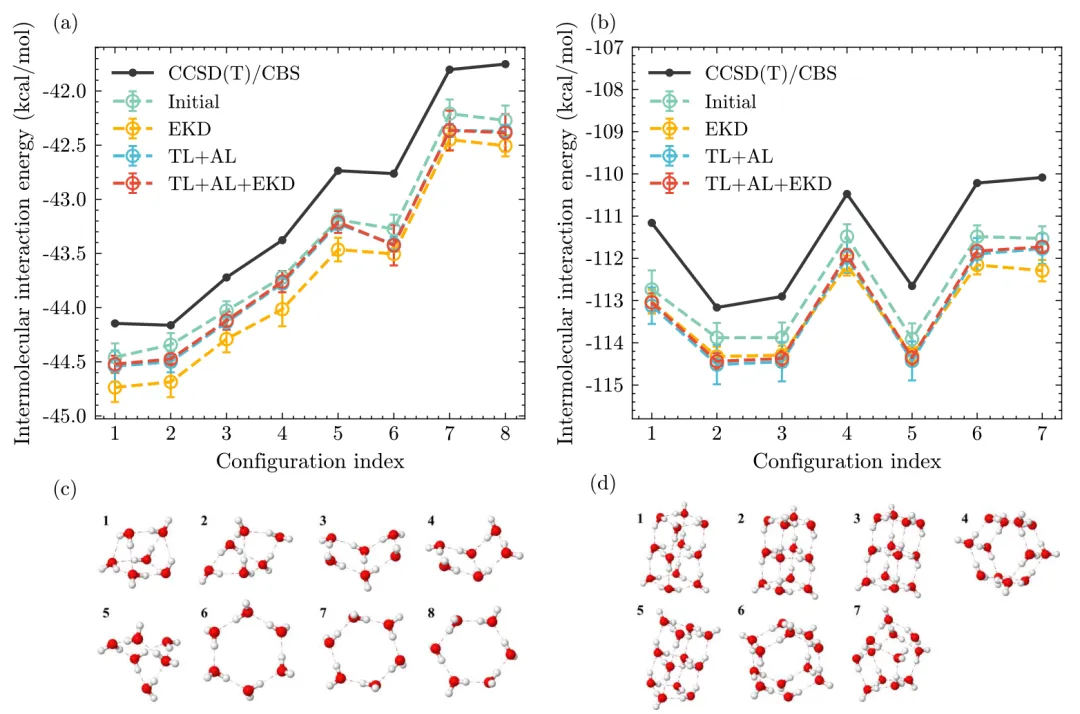
摘要:在前期工作中,我们开发了一种基于 CCSD(T) 精度等级的长程分离型水分子力场,该力场结合了物理驱动模型与机器学习模型的优势。然而研究发现,高昂的 CCSD(T)/CBS 计算成本限制了量子化学(QM)数据集的规模,且由于缺乏力(Force)标签,这两者共同导致了训练的不稳定性问题。体相性质(Bulk properties)表现出的巨大偏差,无法通过简单地降低小团簇 QM 数据集的拟合误差来解决。这种在体相模拟中的不稳定性是机器学习势能(MLPs)训练中的普遍问题,且在基于 CCSD(T) 水平的数据驱动研究中尤为严重。 在本研究中,我们以长程分离水模型为例,旨在通过开发一种全新的训练工作流来克服上述限制。该工作流由以下几项核心技术组成:1. 一种确保在不同温度和密度下实现更彻底采样的主动学习(Active Learning)协议;2. 一种采用机器学习密度泛函的中间力标签技术;3. 一种集成知识蒸馏(EKD)方法。这些技术显著提升了所得水模型的稳定性,使其在团簇能量和实验性质的预测上一致达到了亚化学精度。我们对包括密度、径向分布函数(RDFs)、介电常数、扩散率和红外光谱在内的多种性质进行了基准评估,结果均达到了当前最先进(SOTA)水平,有力证明了该训练协议的有效性。
2 利用半金属型 和 纳米片实现水介导的金、钯、铂回收

英文标题:Water-mediated recycling of gold, palladium and platinum using semimetallic TiS2 and TaS2 nanosheets
作者:Jianhong Wei , Miaofei Huang , Kuang Yu* , Huanjing Liang , Fei Li , Kaiqiang Zheng , Fangluo Chen , Yibo Gao , Yang Su*, Hui-Ming Cheng*
原文链接:https://doi.org/10.1093/nsr/nwaf522
DOI号:10.1093/nsr/nwaf522
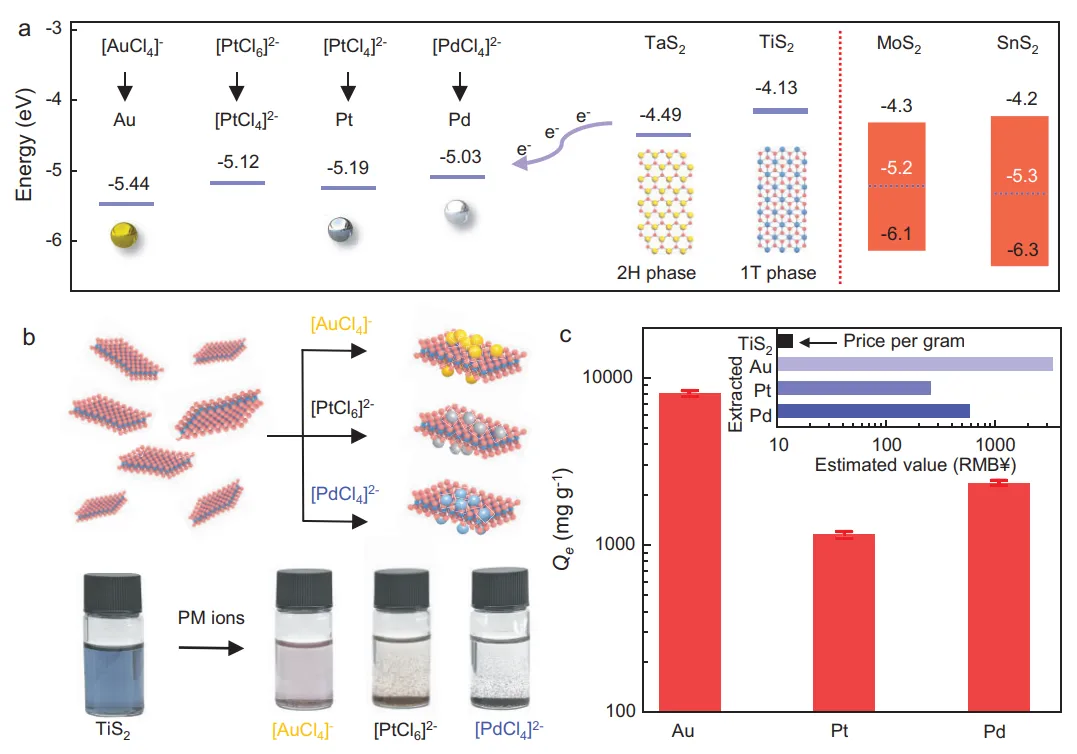
摘要:电子与催化工业中对金 (Au)、钯 (Pd) 和铂 (Pt) 等贵金属(PMs)的大量且不可替代的消耗,加之其在地壳中的稀缺性,迫切需要创新的回收方案以实现贵金属的可持续性。然而,由于在低浓度下萃取容量不足,且研究重心过度集中于金而忽略了其他贵金属,从废弃物浸出液中回收贵金属的努力一直受到阻碍。在此,我们报道了利用半金属型过渡金属硫族化合物—— 和 纳米片,实现对贵金属离子的超高还原性回收及其在水相中的原位沉积。值得注意的是, 对金、钯、铂离子的萃取容量分别达到了前所未有的 ~8、2.3 和 1.15 g/g;吸附的贵金属离子被直接转化为纳米颗粒并沉积在纳米片上。机理研究表明,水介导的电子转移(源自半金属 和 纳米片的硫位点)是实现超高萃取容量的原因,单个 分子可向金离子提供超过 13 个电子。这种电子转移是通过水解离过程中形成的硫-氧物种介导完成的。我们进一步证明了该方法能从电子垃圾、废催化剂和汽车催化转化器等实际废料流中,实现金、钯、铂的选择性及完全回收。
3 用于预测有机液体热导率的加速型机器学习力场

英文标题:Accelerated machine learning force field for predicting thermal conductivity of organic liquids
作者:Wei Feng , Siyuan Liu , Hongyi Wang , Zhenliang Mu , Zhichen Pu , Xu Han, Tianze Zheng, Zhenze Yang,Zhi Wang , Weihao Gao , Yidan Cao , Kuang Yu∗, Sheng Gong∗ , Wen Yan
原文链接:https://doi.org/10.1016/j.mtener.2025.102178
DOI号:10.1016/j.mtener.2025.102178
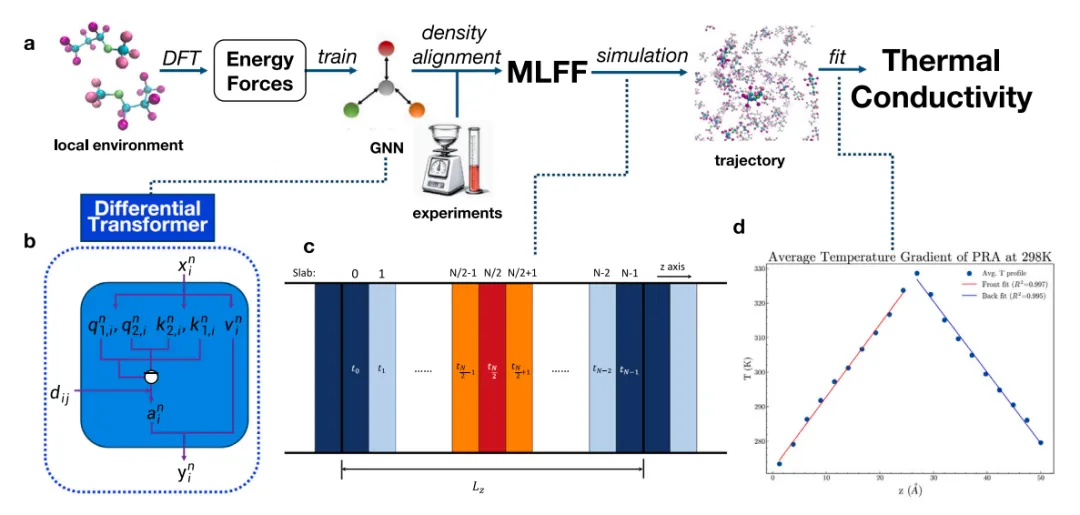
摘要:有机液体的热导率是影响能源转换、电子冷却及化学工艺等各类工业与环境应用的关键参数。然而,有机液体热导率的原子级模拟一直受到双重制约:一是经典力场的精度有限,二是从头算(ab initio)方法高昂的计算需求。 在本研究中,我们提出了一种基于机器学习力场(MLFF)的分子动力学模拟工作流,用于预测 20 种有机液体的热导率。在此,我们将“微分注意力机制”(Differential Attention)引入 MLFF 架构以增强其学习能力,并利用液体密度对 MLFF 进行校准以使其与实验结果保持一致。结果表明,该工作流对多种有机液体热导率预测的平均绝对百分比误差(MAPE)仅为 14%,显著低于目前现有的经典力场(78%)。此外,我们使用 Triton 语言重写了该 MLFF 以最大化模拟速度,从而实现了热导率的快速预测。
4 通过稳定 中间体实现乙醛酸与硝酸盐高选择性电合成甘氨酸

英文标题:Highly Selective Electrosynthesis of Glycine from Glyoxylic Acid and Nitrate via Stabilizing the NH2OH Intermediates
作者:Dr. Weiliang Zhou, Yidi Wu, Wentao Zhang, Zhiyi Chen, Ziyao Gao, Ji Li, Prof. Kuang Yu, Prof. Yan-Bing He, Prof. Bilu Liu, Prof. Feiyu Kang, Prof. Lele Peng*
原文链接: https://doi.org/10.1002/ange.202516749
DOI号: 10.1002/ange.202516749
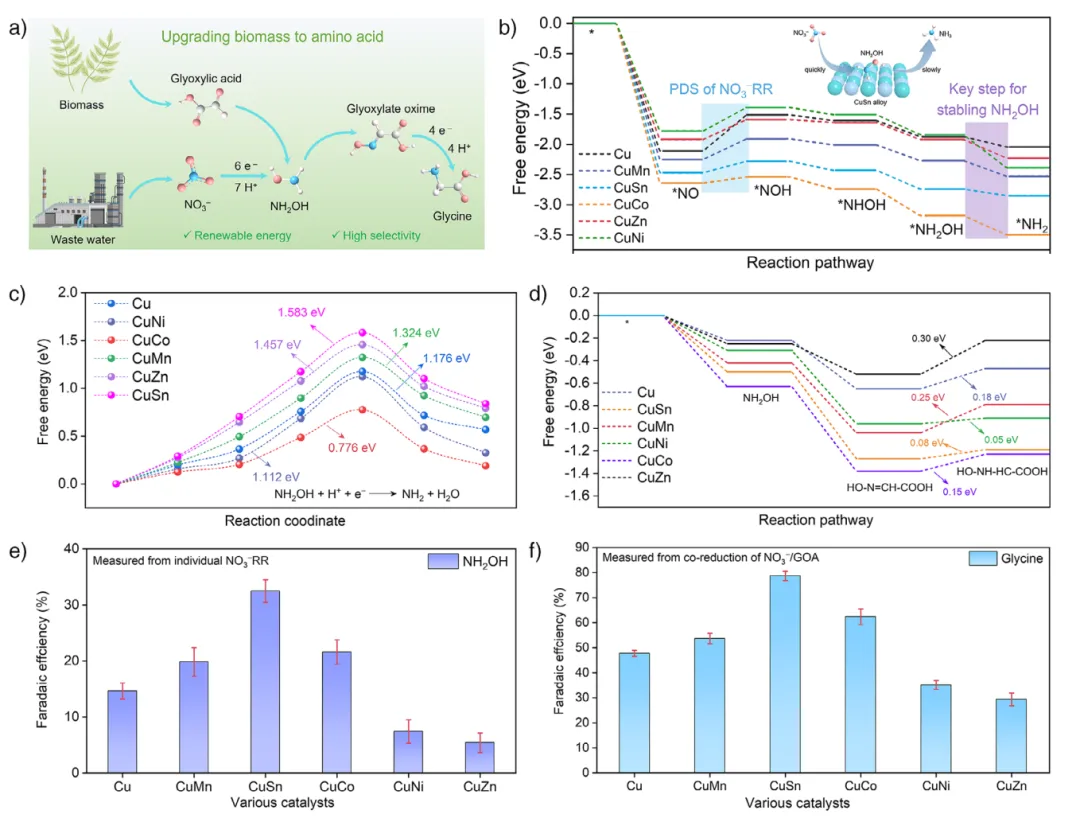
摘要:电化学 C–N 偶联反应已成为在环境条件下合成高附加值有机化合物的重要策略,其中甘氨酸在多种生物活性及药物合成中具有重要的实用价值。然而,由于反应路径复杂,此类合成过程的选择性受到限制。在本研究中,我们报道了通过铜基模型催化剂,利用稳定 中间体并优化乙醛酸吸附的策略,实现了硝酸盐与乙醛酸共还原高选择性合成甘氨酸。理论计算与原位(operando)测量表明,CuSn 合金能促进 的生成并优化乙醛酸的吸附,从而促进 C–N 基团的精准偶联以合成甘氨酸。因此,CuSn 电催化剂在共还原制备甘氨酸中表现出优异性能,其最大法拉第效率和选择性分别达到 78.69% 和 99.32%。此外,在放大合成实验中,CuSn 催化剂能以 100% 的转化率生产 0.73 g 甘氨酸。技术经济分析(TEA)显示,该方法的总成本为 1158.88 美元/吨,比商业价格(2060.44 美元/吨)低 1.78 倍,表明在本系统中电合成甘氨酸是一条可行且盈利的路线。
5 一种用于液体电解质的物理驱动与神经网络混合力场

英文标题:A Hybrid Physics-Driven Neural Network Force Field for Liquid Electrolytes
作者:Junmin Chen*, Qian Gao*, Yange Lin*, Miaofei Huang, Zheng Cheng, Wei Feng, Jianxing Huang, Bo Wang, Kuang Yu
原文链接:https://doi.org/10.48550/arXiv.2511.13294
DOI号:10.48550/arXiv.2511.13294
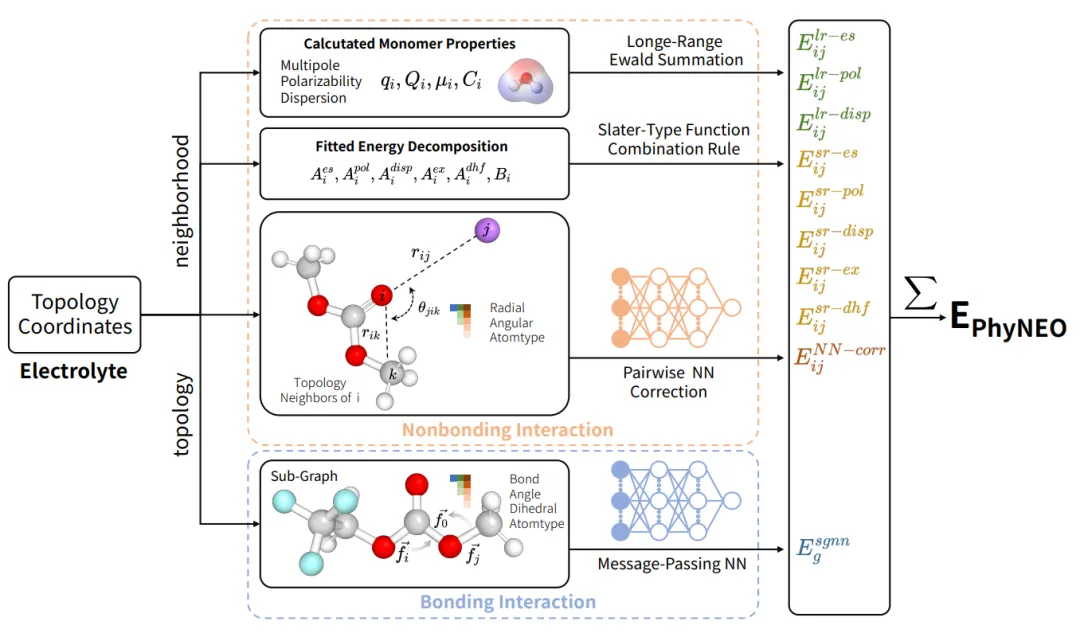
摘要:电解液设计在锂离子电池和钠离子电池的开发中发挥着重要作用。电池电解液拥有巨大的设计空间,由不同的溶剂、添加剂和盐类组成,这使得仅靠实验探索变得十分困难。高保真分子模拟通过采用精确的势能面,能够准确预测电解液的体相性质,从而指导分子设计与配方工程。 目前,过度简化的经典力场严重依赖实验数据进行微调,其在微观层面的预测能力存疑。相比之下,新出现的机器学习原子间势能(MLIP)能准确重现从头算数据,展现出卓越的拟合能力。然而,MLIP 仍受到迁移性低、体相性质预测稳定性不足以及训练成本扩展性差等问题的困扰。因此,它尚不能作为探索电解液设计空间的稳健且通用的工具。 在本研究中,我们引入了一种高度可扩展且完全“自下而上”的力场构建策略,称为 PhyNEO-Electrolyte。它采用物理驱动与数据驱动相结合的混合方法,仅依赖单体和二聚体的能量分解分析(EDA)数据。通过对长/短程作用以及非键/成键相互作用的细致分离,我们严格恢复了长程渐近行为,这对于描述电解液体系至关重要。通过该方法,我们显著提高了 MLIP 训练的数据效率,使我们能够以更少的数据实现更广的化学空间覆盖,同时在体相计算中保持可靠的定量预测能力。因此,PhyNEO-Electrolyte 将成为未来电解液优化的重要工具。
6 用于锌基储能器件锌负极稳定剂的多功能氧化还原活性电解液添加剂

英文标题:Multifunctional redox-active electrolyte additives as Zn anode stabilizers for Zn-based energy storage devices
作者:Mingwu Luo , Wei Feng , Safakath Karuthedath , Kuang Yu , Bo Li , Feiyu Kang* , Mengyao Li*
原文链接:https://doi.org/10.1016/j.cej.2025.170282
DOI号:10.1016/j.cej.2025.170282
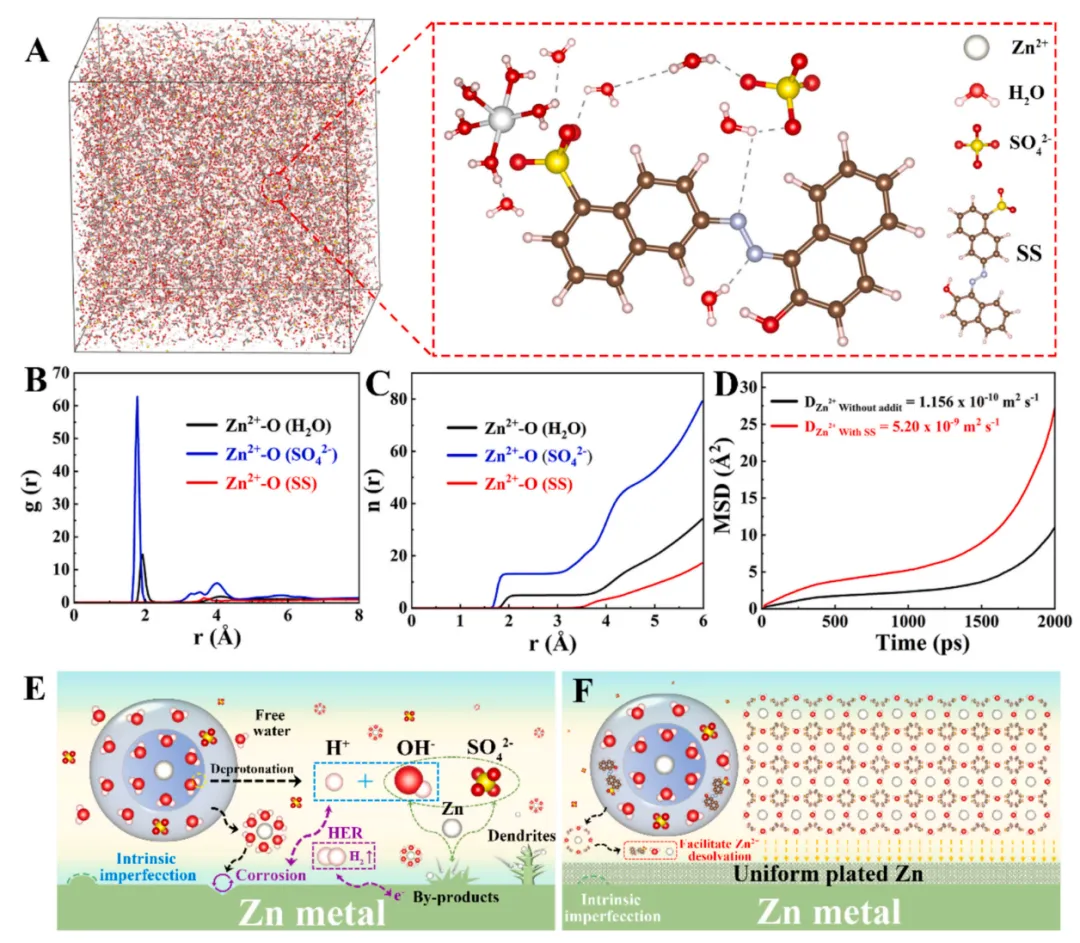
摘要:作为目前主流储能器件(如锂离子电池)更安全的替代方案,水系锌基储能器件(AZDs)正脱颖而出,以满足全球对清洁能源存储与利用不断增长的需求。本研究提出了一种克服金属锌负极集成关键障碍的新方法。通过开发偶氮类氧化还原活性分子作为电解液添加剂,我们证明了通过优化锌-电解液界面,可以同时实现抑制枝晶形成与提升电容性能。计算分析表明,引入的猩红染料(Silk Scarlet)分子通过协同重构水合壳层和配位化学,促进了 溶剂化动力学的优化。该界面工程策略显著改善了循环性能:对称电池在 条件下可保持 2730 小时的稳定性,较无添加剂的电解池寿命大幅提升。此外,这种可逆的氧化还原活性添加剂通过双电子-质子转移介导,加速了 AZDs 中的电荷存储动力学。一系列结构类似分子的实验结果进一步证实了偶氮类电解液改性策略的可靠性。因此,这项工作为利用氧化还原活性添加剂优化 AZD 技术的电解液提供了新途径。
7 基于相图的力场自动精细化方法

英文标题:Automatic Refinement of Force Fields Based on Phase Diagrams
作者:Bin Jin, Bin Han, Wei Feng, Kuang Yu*, Shenzhen Xu*
原文链接:https://doi.org/10.48550/arXiv.2510.16778
DOI号:10.48550/arXiv.2510.16778
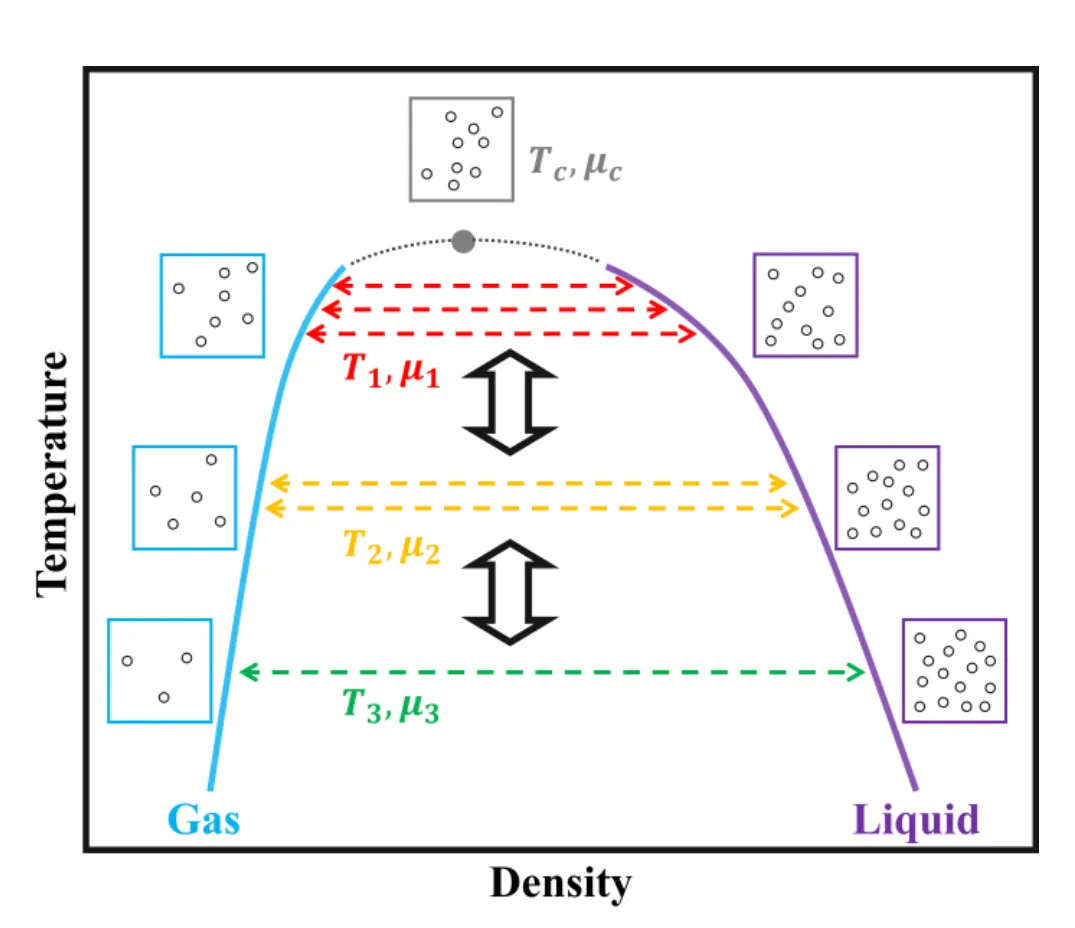
摘要:相变特性的精确表征需要充足的构型采样,这就要求势能面兼具高效性与准确性。分子力场因其计算效率高、物理可解释性强而备受青睐,但针对复杂相互作用对其进行精细化调整仍极具挑战。为了解决这一问题,我们提出了一种以相图作为“自上而下(Top-down)”优化目标,并基于自动微分技术的力场精细化策略。 我们以气液共存性质为范例,采用增强采样技术,并设计了一个可微损失函数(Differentiable loss function)来评估力场对相图的描述精度。精细化后的力场在两种建模体系中生成的气液相图均与目标高度吻合。这证实了我们的方法是一种针对相变研究的高效、自动化力场开发框架。
8 通过 纳米颗粒实现酯类电解液中的溶剂束缚与优化的 溶剂化环境,助力无负极锂金属电池

英文标题:LiNO3 Nanoparticle Enabled Solvent Confinement and a Favorable Li+ Solvation Environment in Ester Electrolytes for Anode-Free Lithium Metal Batteries
作者:Yue Cao,† Guohuang Kang,† Jiachao Duan, Rui Yin, Ying Meng, Kuang Yu,* Feiyu Kang, and Yidan Cao*
原文链接:https://doi.org/10.1021/acsnano.5c09514
DOI号:10.1021/acsnano.5c09514
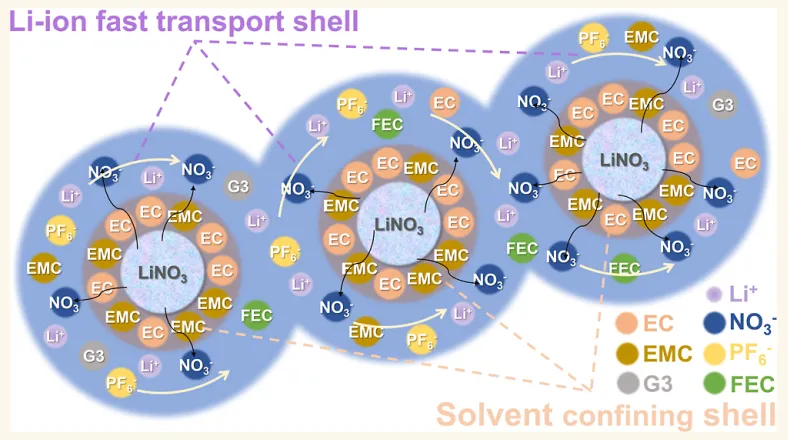
摘要:调控液体电解液中锂离子()的溶剂化环境,对于在锂金属负极上构建稳定的固体电解质界面(SEI)膜至关重要。在本研究中,我们报道了一种通过引入硝酸锂()纳米颗粒作为添加剂,来调节酯类电解液中 溶剂化环境的方法。在 颗粒与电解液界面处的偶极-偶极相互作用(dipole–dipole interactions),导致溶剂分子在 颗粒表面发生有序聚集,形成了一个分子束缚层(molecular confinement layer),进而诱导形成弱 溶剂化环境。这使得 更易与阴离子结合,促进了 的快速传导,并助推了富含无机组分的 SEI 膜的生成。电化学测试表明,由 纳米颗粒引起的这些变化显著提高了库仑效率,降低了锂成核过电位,抑制了锂枝晶生长,并延长了无负极电池的循环寿命。此外,在含有 6000 ppm 水分的电解液中,电池仍实现了超过 200 次的稳定循环,容量保持率为 71.21%。这些发现为先进电解液中固/液界面的溶剂/离子调控提供了见解。
9 通过普适力场连接量子力学与有机液体性质

英文标题:Bridging Quantum Mechanics to Organic Liquid Properties via a Universal Force Field
作者:Tianze Zheng*, Xingyuan Xu, Zhi Wang, Zhenze Yang, Yuanheng Wang, Xu Han, Lei Chen, Zhenliang Mu, Ziqing Zhang, Siyuan Liu, Sheng Gong, Kuang Yu, Wen Yan*
原文链接:https://doi.org/10.48550/arXiv.2508.08575
DOI号:10.48550/arXiv.2508.08575
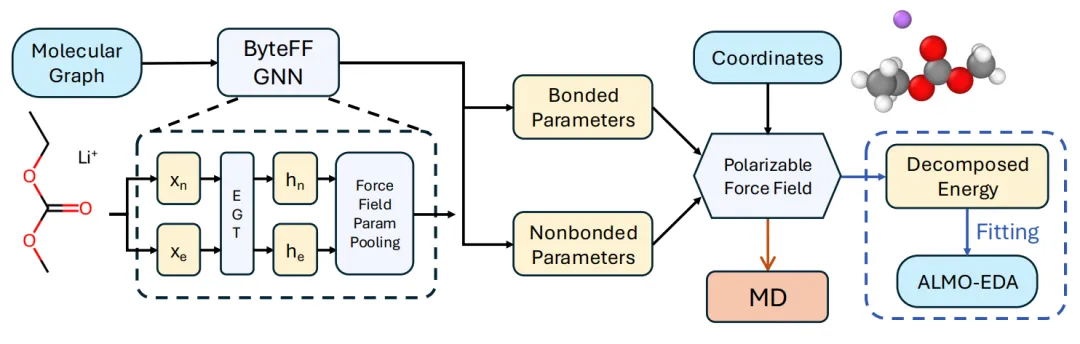
摘要:分子动力学(MD)模拟是揭示凝聚态体系结构与动力学微观机制的重要工具。然而,由于计算成本与模拟精度之间的权衡,从头算(ab initio)方法对宏观性质的普适且准确的预测仍面临巨大挑战。 在此,我们提出了 ByteFF-Pol,这是一种由图神经网络(GNN)参数化的极化力场,完全基于高精度量子力学(QM)数据训练而成。通过利用物理驱动的力场形式和训练策略,ByteFF-Pol 在预测多种小分子液体及电解质的热力学和输运性质方面表现出卓越性能,超越了目前最先进(SOTA)的经典力场和机器学习力场。ByteFF-Pol 的零样本(Zero-shot)预测能力桥接了微观 QM 计算与宏观液体性质之间的鸿沟,使得探索此前难以触及的化学空间成为可能。这一进展对于电解液设计和定制化溶剂开发等应用具有变革性潜力,标志着数据驱动材料发现迈出了关键一步。
10 通过机器学习技术提升聚合物热导率的计算效率与精度

英文标题:Enhancing Thermal Conductivity Computation of Polymers via Machine Learning Techniques
作者:Chengyang Tu, Xin Li, Junmin Chen, Bo Sun, and Kuang Yu*
原文链接:https://doi.org/10.1021/acs.jpcb.5c03656
DOI号:10.1021/acs.jpcb.5c03656
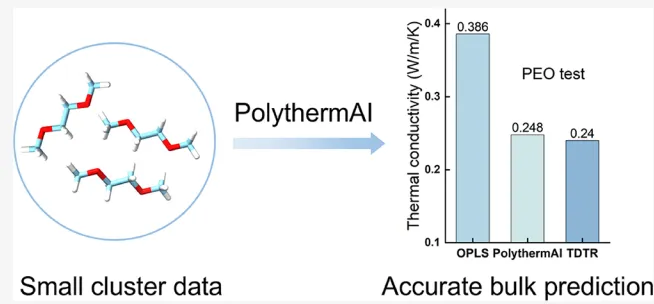
摘要:由于聚合物结构的复杂性,对其热导率()进行准确预测通常极具挑战。目前现有的从头算方法(如 DFT-BTE)计算成本高昂,而分子动力学中使用的经典力场又缺乏足够的精度。在本研究中,我们将从头算杂化机器学习(ML)/多极极化势(即 PhyNEO)与机器学习辅助的热流计算相结合。该方法提供了可靠的热流轨迹,进而用于定量预测聚合物的热导率。我们以聚乙二醇(PEO)为例,将计算结果与通过时域热反射(TDTR)测量获得的可靠实验参考值进行了对比,结果高度吻合。这项工作实现了仅从小团簇量子数据出发即可定量预测块体聚合物的热导率,为未来的广泛应用提供了保障。
11 利用机器学习增强的恒电势框架观测锂金属-电解液界面的枝晶形成

英文标题:Observation of dendrite formation at Li metal-electrolyte interface by a machine-learning enhanced constant potential framework
作者:Taiping Hu, Haichao Huang, Guobing Zhou, Xinyan Wang, Jiaxin Zhu, Zheng Cheng, Fangjia Fu, Xiaoxu Wang, Fuzhi Dai, Kuang Yu* & Shenzhen Xu*
原文链接:https://doi.org/10.1038/s41467-025-62824-5
DOI号:10.1038/s41467-025-62824-5
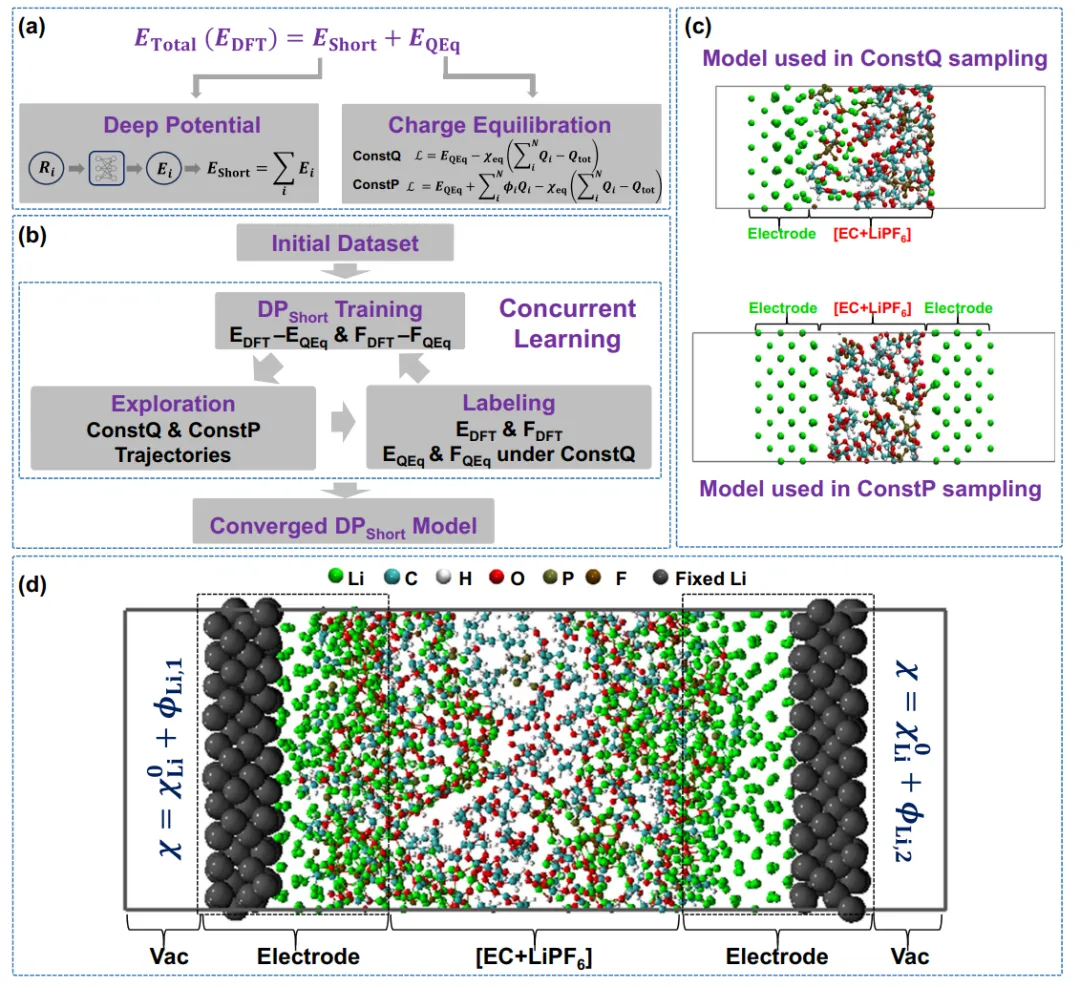
摘要:在电化学循环过程中,不可控的枝晶生长会导致锂金属电池库仑效率低下并引发严重的安全问题。因此,深入理解枝晶形成机制对于进一步提升锂金属电池性能至关重要。 机器学习加速的分子动力学(MD)模拟能够以从头算(ab initio)级别的精度提供各种关键过程的原子尺度分辨率。然而,由于缺乏电化学恒电势条件,传统的分子动力学模拟工具难以捕捉到锂的电化学沉积过程。 在本研究中,我们提出了一种结合了机器学习力场与电荷平衡(Charge Equilibration)方法的恒电势模拟方法,旨在揭示锂金属负极表面枝晶成核的动态过程。我们的模拟表明,在固体电解质界面(SEI)的无定形无机组分中,锂发生聚集并导致非均匀沉积,进而诱发枝晶成核。该研究为锂金属负极中锂枝晶的形成提供了微观见解。更重要的是,我们提出了一种高效且精确的模拟方法来模拟真实的恒电势条件,这在模拟复杂电化学界面方面具有巨大的应用潜力。
12 开启 6,13-双(三异丙基硅烷基乙炔基)并五苯的荧光发射:通过二维材料调控分子堆积以抑制单线态激子裂变

英文标题:Unlocking Photoluminescence in 6,13-Bis(triisopropylsilylethynyl)pentacene: Singlet Fission Mitigation by 2D Material-Controlled Molecular Packing
作者:Zhichao Cheng,∇ Han Zheng,∇ Jing Guo,∇ Qi Wang,∇ Steffen Duhm, Norbert Koch, Hui-Ming Cheng, Kuang Yu,* and Xiaomin Xu*
原文链接:https://doi.org/10.1021/acsnano.5c07029
DOI号:10.1021/acsnano.5c07029
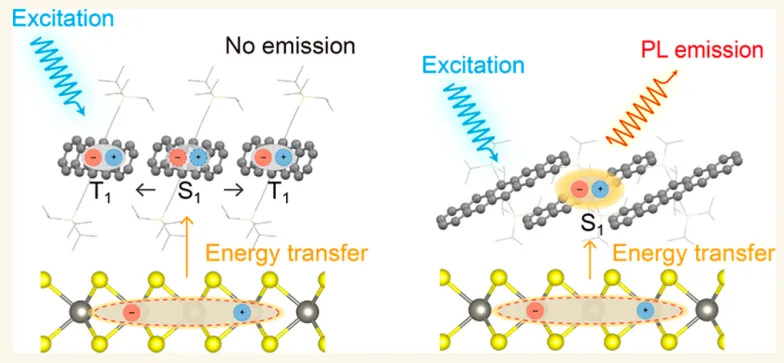
摘要:有机荧光半导体在生物成像、传感及发光器件领域具有巨大的应用潜力。然而,它们在光电器件中的实际应用常受限于聚集诱导淬灭(ACQ)效应,该效应会剧烈降低高密度堆积固体中的荧光(PL)量子产率。在此,我们报道了对 6,13-双(三异丙基硅烷基乙炔基)并五苯(TPn) 分子堆积的调控研究。TPn 是一种典型的有机半导体模型分子,其晶体形式由于存在超快单线态激子裂变,通常表现出可忽略不计的荧光。通过使用单层 (ML-)作为生长衬底,我们诱导了 TPn 界面层分子从“站立式”到“平躺式”取向的转变,从而有效抑制了单线态裂变并开启了其荧光发射。此外,TPn/ML- 异质界面处的 I 型(Type-I)能级对齐结构,通过来自 ML- 的激子能量转移进一步增强了 TPn 的发光。这项工作为利用二维纳米材料定制弱发光 共轭固体的分子堆积提供了一种有效方法,助力其在光电器件中的应用。
13 超强酸辅助的高通量生产与溶液相加工原生二维材

英文标题:Superacid assisted high-throughput production and solution-processing of pristine two-dimensional materials
作者:Qixuan Deng , Jiabin Ma, Minsu Liu* , Yicong Zhou , Siyuan Ding , Ke Zhan , Yuan Zhu d, Zhiyuan Xiong , Yang Su , Kuang Yu* , Hui-Ming Cheng* , Ling Qiu*
原文链接:https://doi.org/10.1016/j.mattod.2025.03.026
DOI号:10.1016/j.mattod.2025.03.026
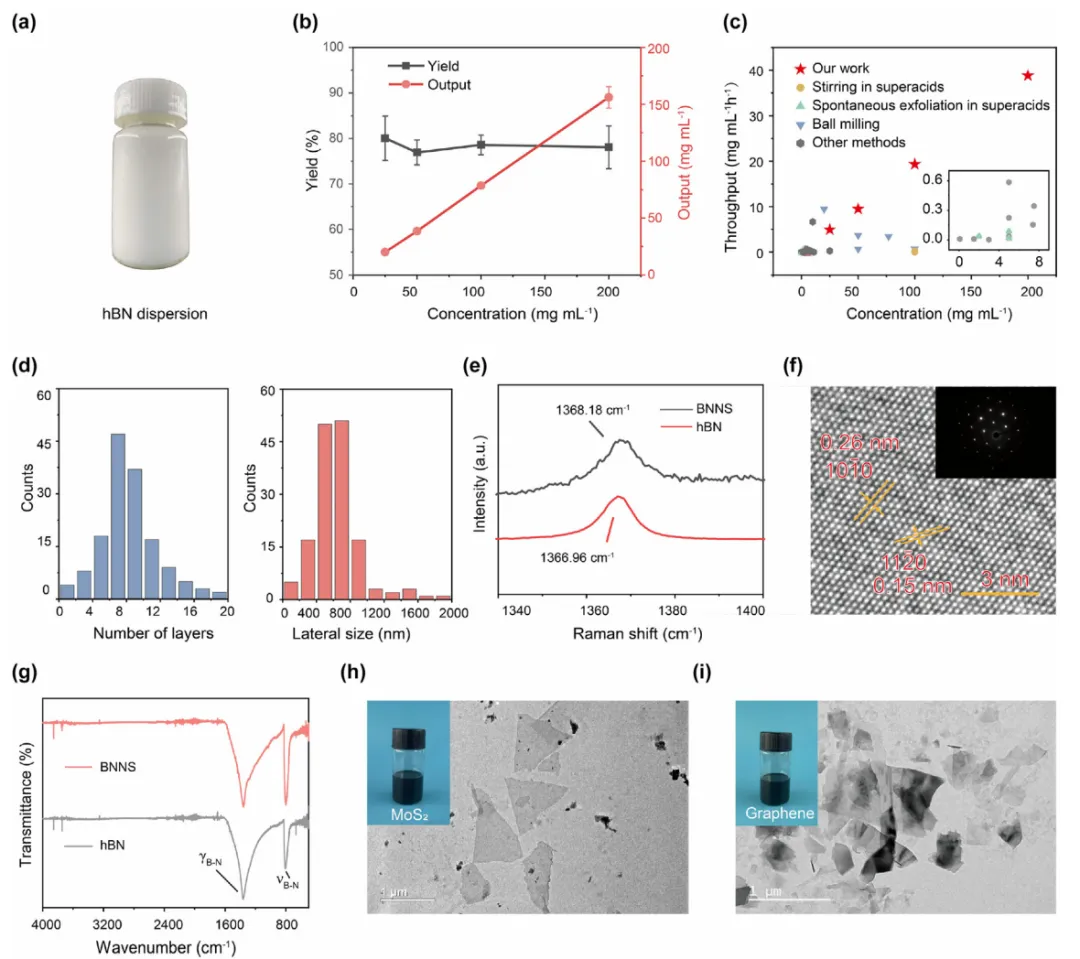
摘要:原生二维(2D)材料具有诸多有趣的特性,拥有广阔的应用前景。然而,由于它们与常用溶剂的亲和力较差且易于聚集,阻碍了其有效的剥离及后续加工成纤维、薄膜等多样化结构,从而限制了其实际应用。在此,我们报道了一种通过在三氟甲磺酸(TfOH)中直接进行高速剪切剥离,从而高效生产原生二维纳米片的方法。典型地,该方法生产六方氮化硼纳米片(BNNSs)的产率达到了前所未有的 38.8 mg mL⁻¹h⁻¹。这种有效的剥离归因于 TfOH 强大的结合亲和力、大的偶极矩以及空间位阻斥力,这些因素共同诱导了六方氮化硼(hBN)的层边缘畸变,从而促进了其剥离。机械剥离与可逆的边缘相互作用不会在剥离后的 BNNSs 上引入额外的官能团。所得的原生 BNNSs 在浓度高达 200 mg mL⁻¹ 的 TfOH 溶液中表现出卓越的稳定性,可保持 28 天以上。通过调节这种高浓度 BNNS/TfOH 分散液的粘度,可以利用相关技术制备出具有高热导率和高机械强度的 BNNS 基球体、纤维和薄膜,适用于先进的热管理应用。室温加工的 BNNS 基球体表现出优于经高温加工的商业化 hBN 或 球体的热导率。此外,TfOH 还可用于辅助石墨烯、二硫化钼和二硫化钨等其他二维纳米片的高效生产与溶液相加工,从而实现高质量大块组装体的制备。
14 论分子力学与机器学习力场之间的设计空间

英文标题:On the design space between molecular mechanics and machine learning force fields
作者:Yuanqing Wang* ; Kenichiro Takaba ; Michael S. Chen; Marcus Wieder ; Yuzhi Xu ; Tong Zhu ; John Z. H. Zhang ; Arnav Nagle; Kuang Yu ; Xinyan Wang ; Daniel J. Cole ; Joshua A. Rackers ; Kyunghyun Cho ; Joe G. Greener ; Peter Eastman ; Stefano Martiniani ; Mark E. Tuckerman
原文链接:https://doi.org/10.1063/5.0237876
DOI号:10.1063/5.0237876
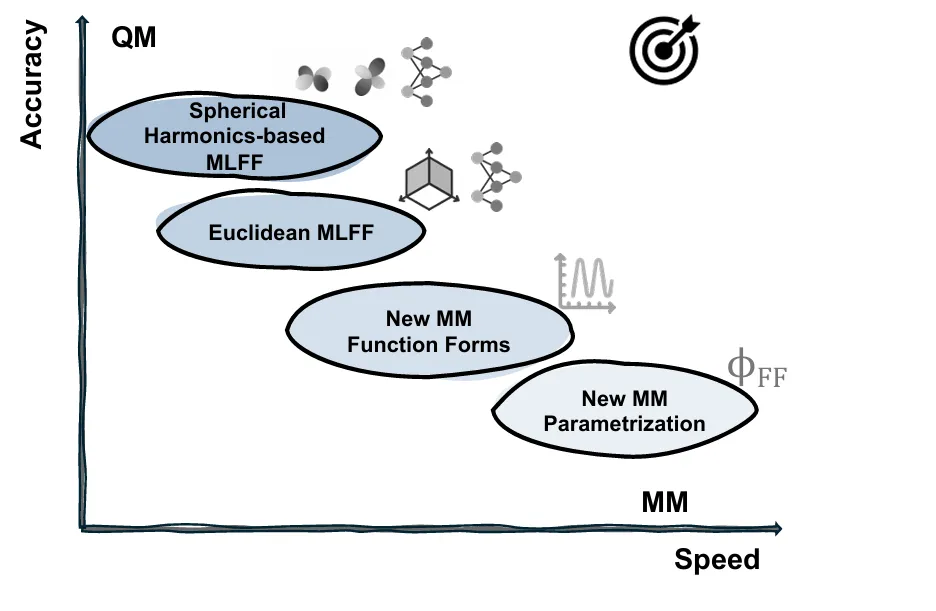
摘要:开发一种既具有量子力学(QM)精度,又具备分子力学(MM)速度的力场,从而能够足够高效且有意义地模拟生物大分子体系并获取定量见解,是生物物理学家最热切的梦想之一——尽管这个梦想在短期内仍难以完全实现。机器学习力场(MLFFs)是朝这一方向迈出的极具意义的一步,它通过自动微分技术,使可微神经函数能够参数化地拟合从头算(ab initio)的能量与受力。 我们认为,就目前而言,MLFF 模型的实用性瓶颈已不再是精度,而主要在于其模拟速度、稳定性以及泛化能力。许多近期的模型变体在受限的化学空间内,早已超越了 1 kcal/mol 的“化学精度”——这是实现可靠化学预测的经验阈值——但其速度仍比 MM 力场慢几个数量级。为了激发开发更快速(尽管精度可能略有下降)的 MLFF 的探索与设计,本综述重点关注了 MM 与 ML 力场之间的技术设计空间(即速度-精度的权衡)。在从机器学习视角简要回顾了这两类力场的构建模块后,我们讨论了当前力场开发群体面临的理想属性与挑战,调研了提升 MM 力场精度以及加快 ML 力场速度的相关努力,并展望了下一代 MLFF 的可能形态。
15 通过无盐型固体聚合物电解质构建富含无机组分的固体电解质界面膜,实现稳定且低成本的锂金属电池

英文标题:Salt-Free Solid Polymer Electrolytes Enabling Inorganic-Rich Solid-Electrolyte Interphase for Stable and Cost-Effective Li-Metal Batteries
作者:Xiangxiang Chen, Junru Wu, Wentao Zhang, Ji Li, Ziyao Gao, Xu Zhao, Kuang Yu, Yan-Bing He, Baohua Li, Feiyu Kang, Lele Peng*
原文链接:https://doi.org/10.1002/smll.202500452
DOI号:10.1002/smll.202500452
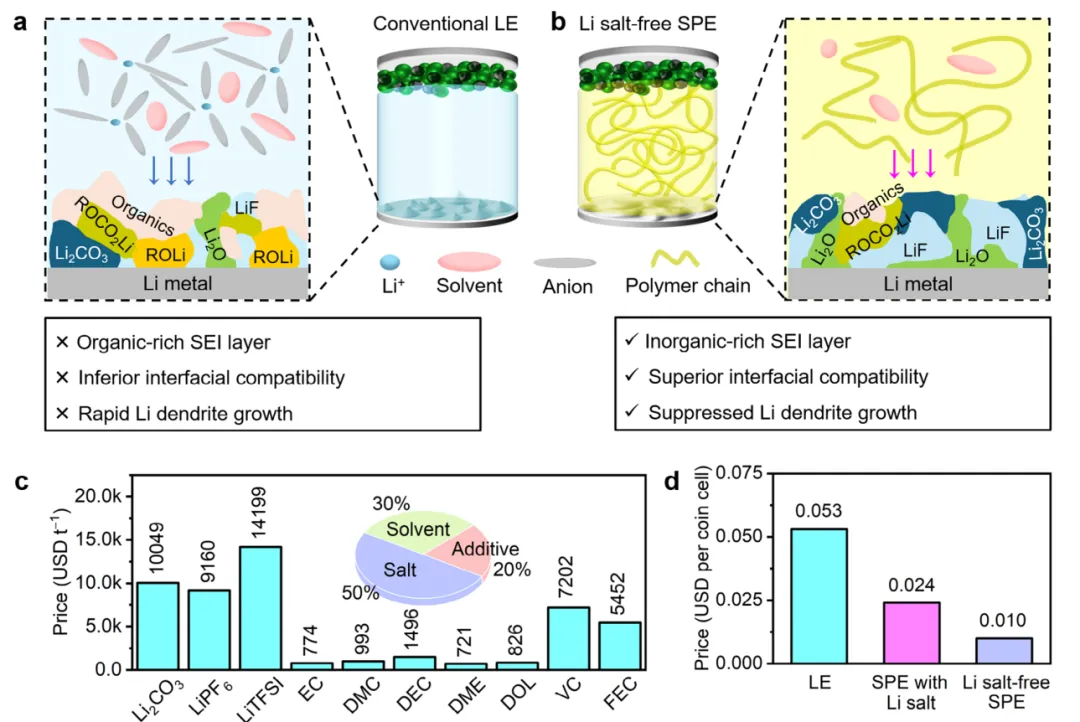
摘要:在电极上构建稳健的固体电解质界面(SEI)膜,是实现液态电解液中锂金属电池稳定运行的关键。然而,液态电解液固有的问题仍无法回避,例如 SEI 的持续腐蚀以及高昂的成本。在此,本文引入了一种新型的无锂盐固体聚合物电解质(SPE),通过 1,3-二氧五环(DOL)和 1,3,5-三氧杂环己烷(TO)在添加离子液体后的原位聚合而成,旨在消除液态电解液的缺陷。聚合物与离子液体之间的均匀相互作用构建了协同的 传导路径,促进了 从正/负极中的脱嵌以及在整个聚合物网络中的顺畅传输,从而赋予了该体系比传统液态电解液更高的 迁移数(0.63)以及可比的离子电导率(1.21 mS cm⁻¹)(传统液态电解液分别为 0.45 和 5.51 mS cm⁻¹)。锂盐的缺失防止了电解液中锂盐氧化分解产生有害且具腐蚀性的酸性物种。更引人注目的是,通过离子液体与聚合物基体的结合,可以实现阴离子占主导的溶剂化构型,进而诱导在电极上形成富含无机组分且抗腐蚀的 SEI 膜。所得的无盐型 SPE 使锂||磷酸铁锂(Li||LiFePO4)电池表现出卓越的循环稳定性,在 780 次循环后容量保持率仍超过 92%。
16 基于分子模拟多模态优化的水系锌电池电解液筛选与设计

英文标题:Screening and Design of Aqueous Zinc Battery Electrolytes Based on the Multimodal Optimization of Molecular Simulation
作者:Wei Feng,∇ Luyan Zhang,∇ Yaobo Cheng, Jin Wu, Chunguang Wei, Junwei Zhang, and Kuang Yu*
原文链接:https://doi.org/10.1021/acs.jpclett.5c00341
DOI号:10.1021/acs.jpclett.5c00341
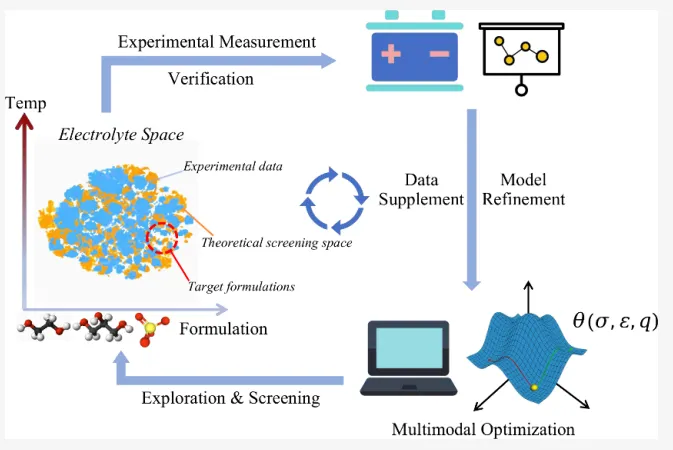
摘要:水系电池,如水系锌离子电池(AZIB),因其本质安全、低成本和环境友好等优势而备受关注。然而,水系电解液在低温下易结冰,限制了其潜在的工业应用。因此,水系电解液设计的核心挑战之一是优化配方,在防止结冰的同时保持良好的离子导通性。然而,传统的实验试错法效率低下,而现有的模拟工具对于高通量相变预测要么不够准确,要么计算成本过高。 在本研究中,我们利用少量实验数据和微分模拟技术开发了一种多模态优化工作流。在极少人工干预的情况下,该工作流显著增强了经典力场对电导率的预测能力。最重要的是,模拟得到的电导率可以作为电解液低温结冰情况的有效预测指标。总的来说,本研究开发的工作流为电解液设计引入了一种新范式。该范式结合了易于测量的实验数据和快速模拟技术,能够预测仅凭单一方法难以获取的复杂性质。
17 迈向六方氮化硼中单光子发射器电子结构的精确表征

英文标题:Towards an accurate electronic structure of single photon emitters in hexagonal boron nitride
作者:Yilin Chen, Haoxiang Chen, Nikolay Bogdanov, Kuang Yu , Ali Alavi, Enge Wang*, and Ji Chen
原文链接:https://doi.org/10.1103/PhysRevResearch.7.L012079
DOI号: 10.1103/PhysRevResearch.7.L012079
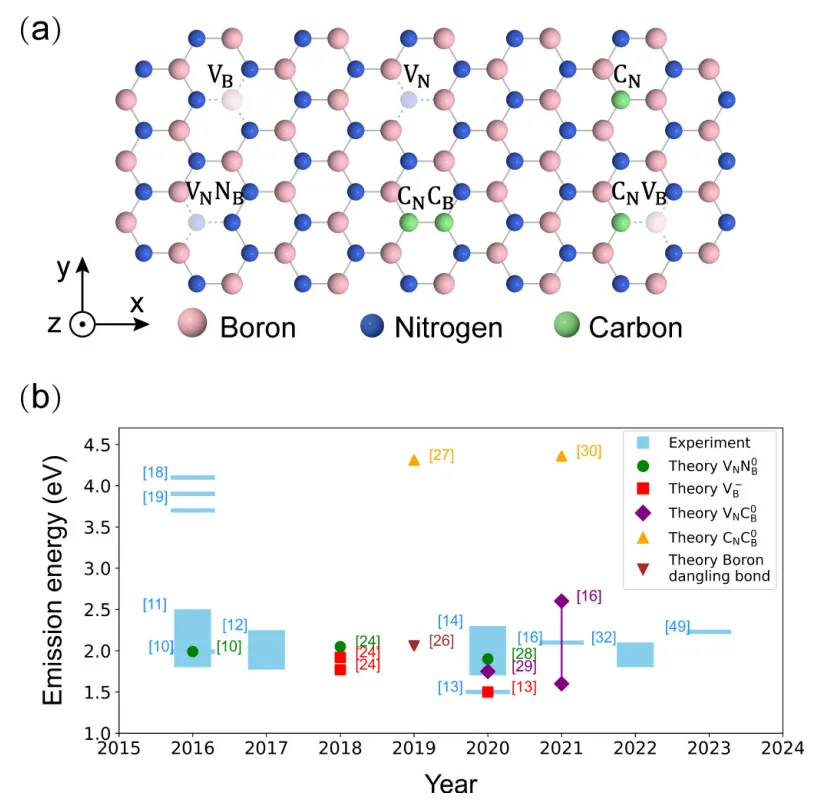
摘要:理解六方氮化硼(h-BN)缺陷中的单光子发射,是将该材料进一步应用于室温量子器件的关键且具挑战性的一步。在此,我们利用全构型相互作用量子蒙特卡罗(FCIQMC)方法,系统地考察了 h-BN 中潜在的发光候选缺陷。 通过这种对电子相关作用的高能级处理,我们最终确定了所有研究缺陷的电子态,揭示了可用于量子操纵的高自旋基态,并纠正了此前的误解。此外,通过计算低能激发态并分析相应的多体波函数,我们确定了可见光区神秘发光带的两个潜在来源,其中包括一个双重态-双重态(doublet-doublet)跃迁。我们的计算阐明了 h-BN 中单光子发射器复杂的关联电子态,并为解决电子结构之外的其他挑战奠定了可靠的理论基础。
18 通过可微分子模拟利用动力学性质精细化势能面

英文标题:Refining potential energy surface through dynamical properties via differentiable molecular simulation
作者:Bin Han & Kuang Yu*
原文链接:https://doi.org/10.1038/s41467-025-56061-z
DOI号:10.1038/s41467-025-56061-z
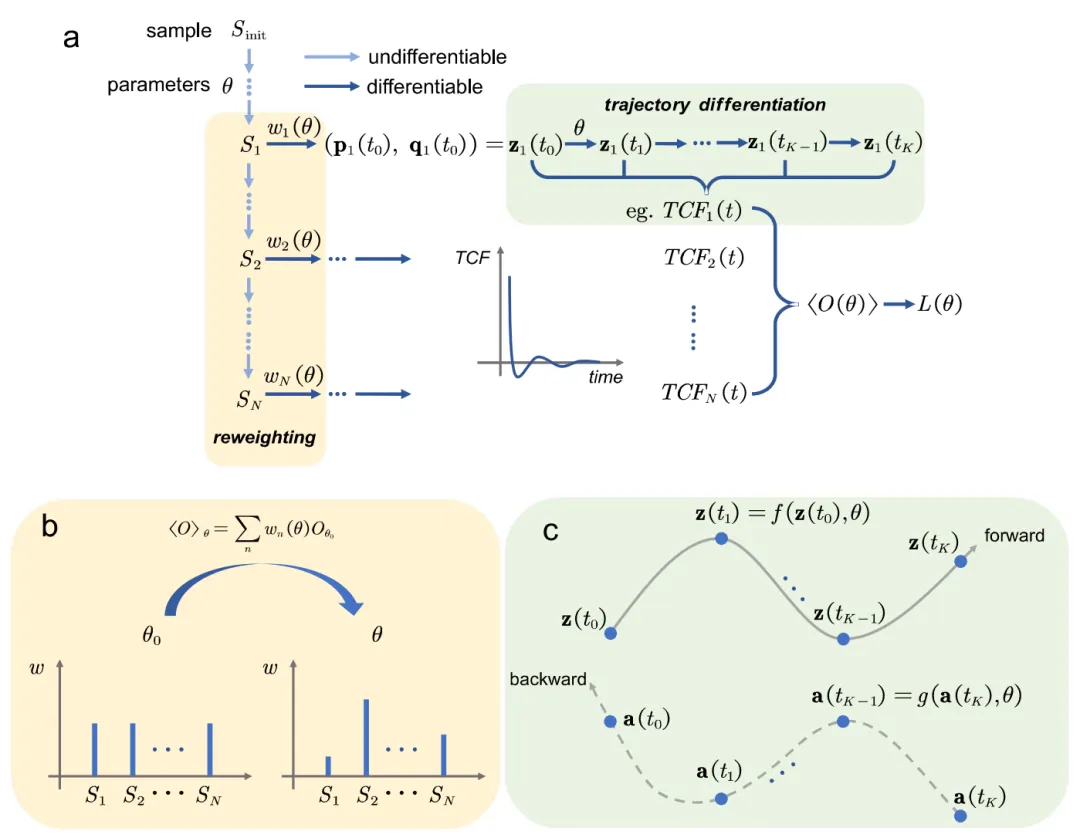
摘要:近年来,机器学习势(MLP)极大地增强了分子动力学模拟的可靠性,但其精度仍受限于底层的从头算(ab initio)方法。克服这一局限的一种可行方案是通过学习实验数据来精细化势能面,得益于现代自动微分技术,这一过程现已能高效实现。然而,目前的势能面精细化大多利用热力学性质进行,使得最易获取且包含丰富信息的动力学数据(如光谱)未被充分利用。 在本研究中,通过综合应用伴随方法(Adjoint method)和梯度裁剪(Gradient truncation)技术,我们证明了在多种情境下均可规避内存溢出和梯度爆炸问题,从而使动力学性质的微分过程保持良好的数学特性。因此,输运系数和光谱数据均可被用于提升基于密度泛函理论(DFT)的机器学习势的精度。本质上,这项工作通过从振动光谱数据中提取微观相互作用,为解决“光谱反演问题(Inverse problem of spectroscopy)”做出了贡献。
19 基于自动微分的恒电势模型高效参数化与部署:在模拟异质 M–N–C 催化电极中的应用

英文标题:Efficient parametrization and deployment of constant potential models based on automatic differentiation: application for simulating heterogeneous M–N–C catalytic electrodes
作者:Haichao Huang, Taiping Hu, Jiaxin Zhu, Shenzhen Xu and Kuang Yu*
原文链接:https://doi.org/10.1039/D5CP02501J
DOI号:10.1039/D5CP02501J
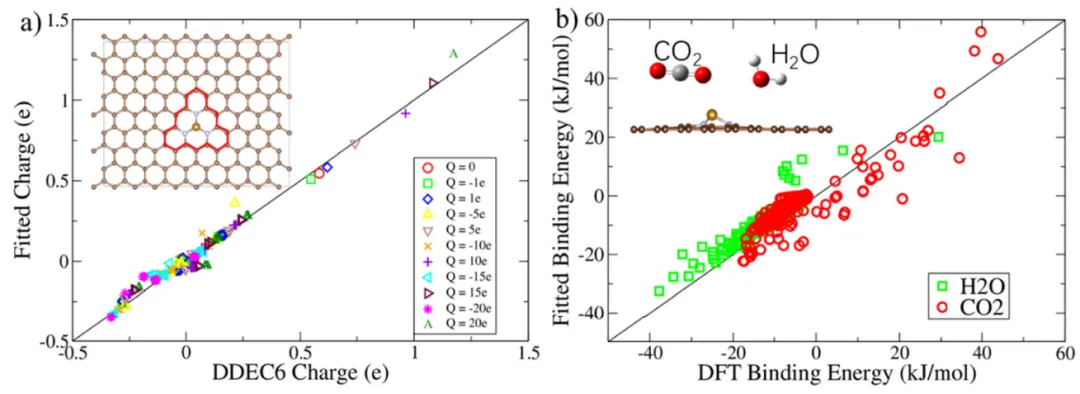
摘要:在本研究中,我们提出了一种集成在基于 JAX 的可微分子力场(DMFF)软件包中的可微电荷平衡(Qeq)方法。这一创新实现了各种性质对原子 Qeq 参数的高效自动微分,促进了针对电化学催化界面构建通用的力场优化与模拟框架。 我们以 M–N–C 催化剂作为模型体系,研究了其在水系电解液中的 CO2 吸附行为。研究结果表明,M–N–C 材料的表面电荷主导了界面水分子的取向和密度分布,从而形成了截然不同的水合层结构。值得注意的是,带负电的电极最有利于掺杂中心对 CO2 的吸附,因为它在静电吸引力与水分子的竞争吸附之间达到了最佳平衡。这些观测结果为 CO2 还原催化剂的合理设计提供了关键见解,并证明了在电化学界面模拟中引入显式溶剂(Explicit solvent)的重要性。
20 现代人工智能技术在有机分子力场开发中的应用

英文标题:Application of modern artificial intelligence techniques in the development of organic molecular force fields
作者:Junmin Chen, Qian Gao, Miaofei Huang and Kuang Yu *
原文链接: https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp02989e
DOI号: 10.1039/D4CP02989E
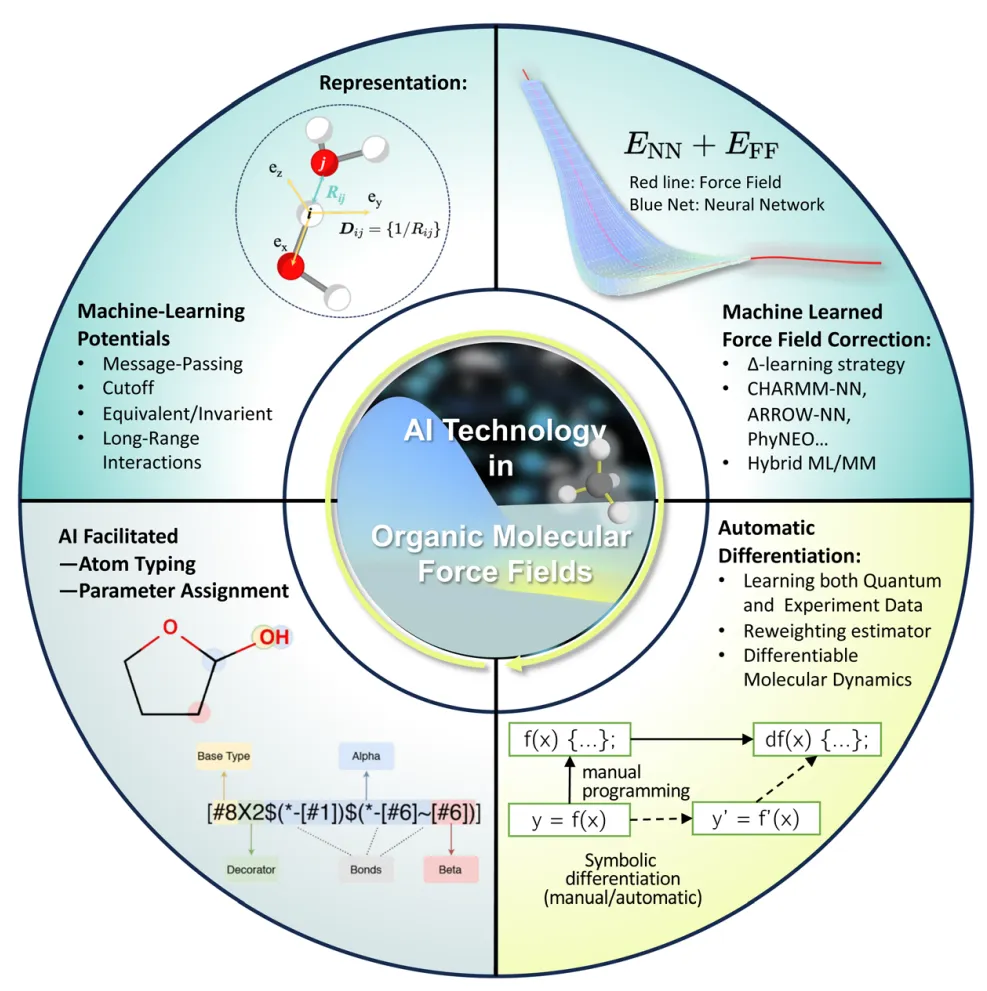
摘要:分子力场(FF)决定了分子动力学(MD)模拟的准确性,是限制 MD 在分子设计中应用的主要瓶颈之一。近年来,人工智能(AI)技术,如机器学习势(MLPs),正在迅速重塑 MD 的研究格局。与此同时,有机分子体系具有独特的特征,在模型构建、优化及验证方面都需要更加细致的处理。虽然目前仍缺乏一种既准确又通用的有机分子力场,但在 AI 的助力下,该领域已取得显著进展,预示着充满希望的前景。 在本综述中,我们概述了用于分子力场开发的各类 AI 技术,并讨论了这些方法的优劣。我们展示了 AI 方法在势能拟合、原子类型化(Atom typification)以及自动优化等诸多任务中表现出的前所未有的能力。同时,值得注意的是,仍需在提升模型迁移率(Transferability)、建立更完善的数据库以及制定更标准化的验证流程方面付出更多努力。通过这些讨论,我们希望激励更多研究投入到解决现有问题中,最终促成下一代通用有机力场的诞生。
21 通过机器学习分子动力学模拟探索体心立方(BCC)镁-锂-铝(Mg-Li-Al)合金的塑性变形机制

英文标题:Exploring the mechanism of plastic deformation in BCC Mg-Li-Al alloys via Machine learning Molecular dynamics simulations
作者:Gaoshang Zhang , Wei Mei , Wentao Zhang , Kuang Yu*
原文链接:https://doi.org/10.1016/j.commatsci.2024.113396
DOI号:10.1016/j.commatsci.2024.113396
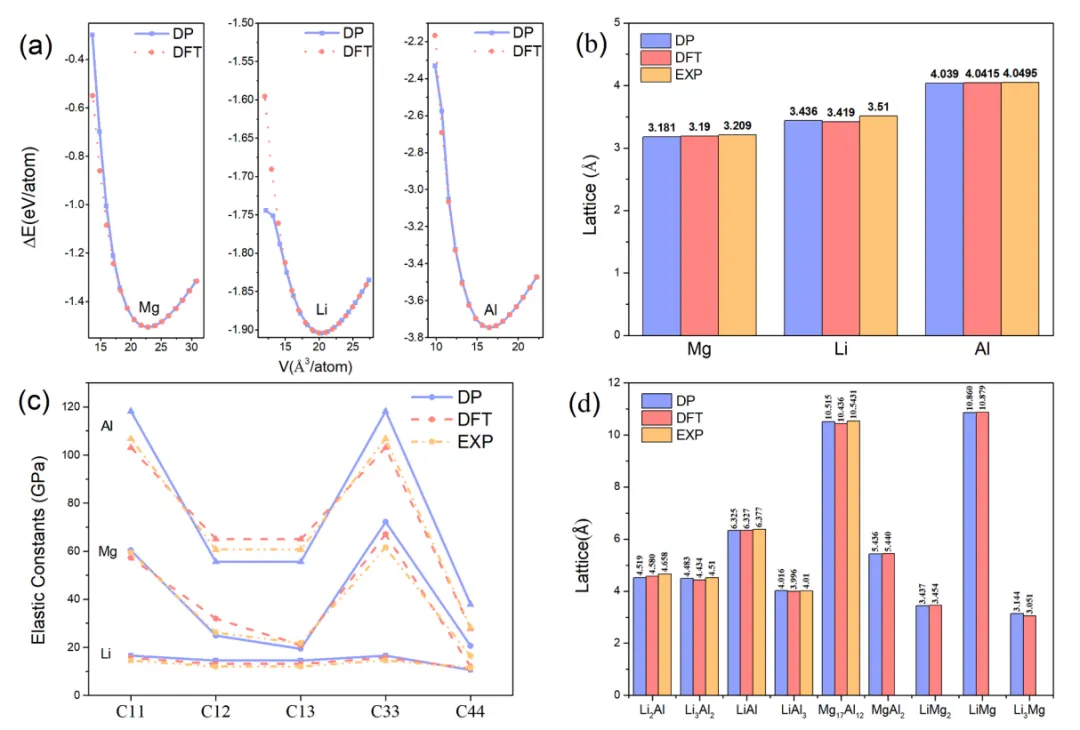
摘要:合金处于材料科学的前沿,其卓越的力学性能对于各种工业应用至关重要。然而,理解合金复杂的塑性行为仍是一项挑战。本研究深入探讨了体心立方(BCC)结构镁-锂-铝(Mg-Li-Al)合金的塑性变形机制,这是一种重要的超轻质结构材料。 我们基于第一性原理计算构建了精确的机器学习势能面,使我们能够在原子尺度上观测单晶和多晶 Mg-Li-Al 合金的塑性变形。我们的模拟揭示了单晶 Mg-14Li-3Al 合金中存在一种复杂且独特的 BCC-FCC-BCC-HCP 马氏体相变过程,这种转变在大拉伸应变下产生了强烈的孪晶强化效应。这种效应在多晶 Mg-Li-Al 中并不存在,但有望被利用于开发新型超强轻质合金。对于多晶体系,我们系统地预测了不同成分比例下的塑性强度,为该合金的进一步优化提供了有用的指导。通过本研究,我们展示了在机器学习势能面的辅助下,现在可以更加便捷地从微观层面理解塑性行为,从而为合金材料的设计开启了新的大门。
通讯作者

职称特任研究员,副教授
主要研究方向为理论化学与计算材料学。主要采用物理模型和数值计算在原子分子尺度上模拟材料体系的微观行为。对复杂的实验现象的微观机制进行理解,并对材料体系的性质进行预测。具体而言,研究主要围绕分子力学模拟方法的发展与应用,以及多尺度电子结构理论开展。主要研究方向包括:1.完全基于第一性原理的精确分子力场模型的发展;2.新型柔性多孔材料(包括MOF、COF等)的结构以及其吸附性能的模拟研究;3.无序固体中热输运现象的模拟,尤其是针对其中量子效应的理论研究;4.量子嵌入理论的发展。
2004-2008年:北京大学,化学与分子工程学院,学士
2008-2013年:美国威斯康星大学麦迪逊分校,化学系,博士
2013-2016年:美国普林斯顿大学,机械与航天工程系,博士后
2016-2018年:美国纽约D. E. Shaw Research公司,研究科学家
2018年至2021年:清华-伯克利深圳学院,清华大学深圳国际研究生院,助理教授
2022年:清华大学深圳国际研究生院,副教授
推荐阅读:
为何99%的单原子催化剂在电池里“躺平”?中科大葛君杰/姜政AM揭示关键:错位的三相界面
中科大JACS葛君杰教授团队重磅突破:让铁原子“高能旋转”,燃料电池阴极效率飙升的秘密
从实验室到真实器件!长春应化所邢巍/肖梅玲JACS全链条验证"超耐久"低铂燃料电池催化剂!
中科大JACS葛君杰教授团队重磅突破:让铁原子“高能旋转”,燃料电池阴极效率飙升的秘密
把水变氧气太浪费?天大巩金龙团队nc巧设“岗位”,让水在电极上专心制造万亿市场化学品!
整理:L
审核:嘻歪歪


