清华大学深圳国际研究生院干林等Adv. Mater.丨动态金属-氧化物界面调控:破解燃料电池催化剂活性-稳定性难题
文章信息标题:Harnessing Dynamic Metal-Oxide Interfaces for Durably Active Fuel Cell Electrocatalysis
作者:Yuefei Cui, Liang Chang, Xiangyu You, Xuan Luo, Wenting Cui, Zejian Li, Sijie Wang, Mengnan Yan, Guilherme V. Fortunato, Marc Ledendecker, Lin Gan
通讯作者:Marc Ledendecker,慕尼黑工业大学,Email: marc.ledendecker@tum.de
干林(Lin Gan),清华大学深圳国际研究生院,Email: lgan@sz.tsinghua.edu.cn
单位:清华大学深圳国际研究生院、慕尼黑工业大学期刊:Advanced Materials (2026)
https://doi.org/10.1002/adma.202519886

一、研究背景
质子交换膜燃料电池(PEMFCs)是实现氢能高效利用的关键技术,但其商业化进程长期受制于阴极氧还原反应(ORR)的缓慢动力学。铂(Pt)基催化剂是目前最有效的ORR催化剂,但面临着活性与稳定性的根本性矛盾:
- 活性提升:通过形貌控制(如八面体PtNi纳米晶)、合金化(PtCo、PtFe)等策略,可将Pt的ORR质量活性提升近一个数量级。然而,这些高活性催化剂通常颗粒较大、电化学表面积(ECSA)较低,导致在高电流密度区(HCD,>1 A cm⁻²)的氧气传质阻力剧增,实际膜电极(MEA)性能并无优势。
- 稳定性挑战:为提升HCD性能,必须减小Pt颗粒尺寸、提高Pt载量(>50 wt%),但这会加剧Pt的溶解、迁移、奥斯特瓦尔德熟化,导致催化剂快速失活。
金属-氧化物界面已被证明可显著调控(电)催化性能。例如,FeO/Pt(111)界面在CO氧化中展现出独特的界面协同效应。然而,这些研究多在理想气相条件下进行,对电化学工况下(尤其是PEMFC实际运行电位区间)界面的动态演化认识严重不足。更重要的是,能否利用金属-氧化物界面的动态行为,在原子尺度上解耦活性与稳定性的矛盾,仍是未解之谜。
二、全文速览
本研究以p区金属氧化物(MOx, M = In, Sn, Sb)修饰的Pt八面体纳米晶为模型催化剂,首次揭示了MOx/Pt界面在PEMFC阴极ORR电位区间内的动态“呼吸”行为:
- 低电位(0.05–0.6 V vs. RHE,模拟工作电位):MOx发生部分还原,界面氧原子脱出,形成氧缺陷的Pt–M金属键界面。该界面通过电荷转移(M→Pt)调变Pt电子结构,削弱含氧中间体吸附,显著提升ORR本征活性。活性增强顺序与M–Pt电负性差一致:In > Sn > Sb。
- 高电位(0.6–1.0 V vs. RHE,模拟启动/怠速/空载电位):MOx保持氧化态,形成富氧的Pt–O–M界面。该界面通过强金属-载体相互作用(SMSI)有效抑制Pt的溶解与迁移,稳定性提升顺序与氧化物晶格匹配度相关:SnOx(金红石相,与Pt(111)晶格失配仅4.7%)> InOx(立方相,失配21.1%)> SbOx(非晶)。
关键突破:将活性最优的InOx与稳定性最优的SnOx复合,制备InSnOx/Pt催化剂,在保持高活性的同时,ECSA与质量活性在加速老化测试后几乎无衰减。进一步将这一策略拓展至PtCo金属间化合物催化剂,InSnOx/PtCo/C在MEA测试中展现出三倍于原始PtCo/C的稳定性,且HCD性能显著提升,达到当前文献报道的顶尖水平。
核心结论:动态金属-氧化物界面可随电位“呼吸”切换构型,在活性与稳定性之间实现原子尺度的动态平衡,为燃料电池催化剂设计提供了全新的普适性策略。
三、文章亮点
- 首次揭示MOx/Pt界面的电位依赖“呼吸”行为结合像差校正HAADF-STEM、iDPC/dDPC成像与operando ICP-MS,在原子尺度上直接观测到Pt–O–M ↔ Pt–M的可逆结构转变,并建立界面构型-电位-催化性能的直接关联。
- 解耦活性与稳定性的原子机制
- 活性源于低电位下形成的Pt–M金属键:电负性差驱动电子从M向Pt转移,下调Pt d带中心,削弱含氧物种吸附(In效果最佳);
- 稳定性源于高电位下形成的Pt–O–M界面:强SMSI效应锚定Pt原子,抑制溶解与迁移(SnOx晶格匹配最佳)。
- “活性-稳定性”协同优化新范式将In(活性剂)与Sn(稳定剂)复合,制备InSnOx/Pt催化剂,实现1+1>2的协同效应:活性媲美InOx/Pt,稳定性媲美SnOx/Pt。
- 从模型催化剂到实用MEA的全链条验证策略成功拓展至高载量Pt/C(50 wt%)和PtCo金属间化合物催化剂,在H₂-空气MEA测试中展现出优异的HCD性能与超长耐久性(30,000圈方波ADT后性能仍保持领先)。
四、图文解析
图1:MOx/八面体Pt模型催化剂的构建与ORR性能
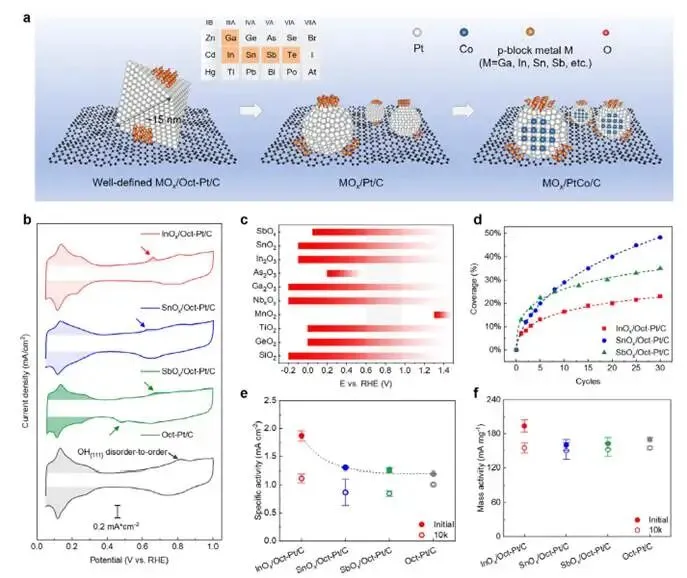
图1a:示意图展示两类催化剂:
- 左:八面体Pt纳米晶(Oct-Pt),暴露(111)晶面,用于原子尺度界面机制研究;
- 右:非晶形Pt/C及PtCo/C,用于验证策略普适性。
图1b:循环伏安(CV)曲线确认MOx沉积
- 在含0.01 mM In³⁺、Sn²⁺、Sb³⁺的0.1 M HClO₄中电位循环沉积;
- InOx/Pt出现In的氧化还原峰(~0.6 V),SnOx/Pt出现Sn²⁺/Sn⁴⁺特征峰,SbOx/Pt出现Sb的氧化峰;
- 通过循环圈数精确控制MOx覆盖率(~15%为最优)。
图1c:p区金属氧化物在PEMFC工况(pH ≤4)下的热力学稳定电位窗口(Pourbaix图):
- In₂O₃、SnO₂、Sb₂O₅在0–1.0 V区间内热力学稳定,适合作为界面修饰层。
图1d:MOx覆盖率随沉积圈数的变化
- InOx沉积最快,3圈达~15%;SnOx次之,5圈达~15%;SbOx最慢,3圈仅~10%。
图1e–f:ORR活性(ADT前后)对比
- 初始活性:InOx/Oct-Pt/C > SnOx/Oct-Pt/C > SbOx/Oct-Pt/C > Oct-Pt/C;
- ADT后:SnOx/Oct-Pt/C和SbOx/Oct-Pt/C活性几乎不变,InOx/Oct-Pt/C活性下降,Oct-Pt/C活性大幅衰减;
- 活性顺序与电负性差Δχ_Pt-M正相关(图S5f):In(0.4)> Sn(0.32)> Sb(0.23);
- CO stripping(图S5g):InOx/Pt的CO氧化峰负移最多,表明界面促进OH吸附,增强CO氧化能力。
图2:像差校正STEM揭示MOx/Pt界面原子结构
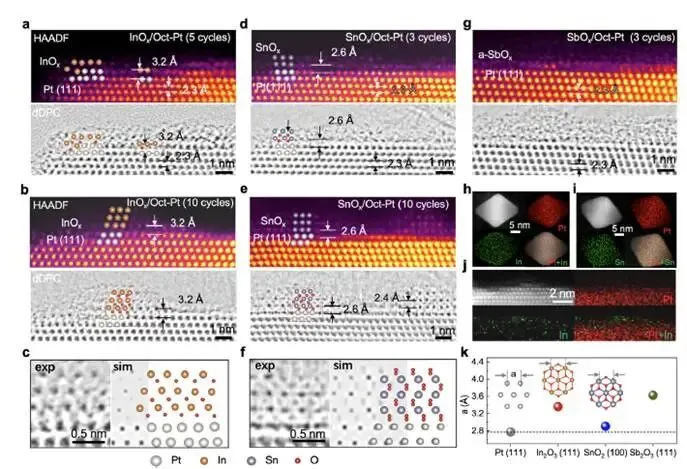
图2a–c:InOx/Oct-Pt界面
- 5圈沉积(图2a):HAADF和dDPC图像显示,1–2原子层厚的InOx不连续覆盖于Pt(111)表面;
- 界面氧原子层清晰可见(dDPC),形成In–O–Pt结构;
- In–Pt层间距3.2 Å,大于Pt(111)面间距(2.3 Å);
- 10圈沉积(图2b):InOx增厚至3–5层,部分区域外延生长,取向{111}<011>In₂O₃//{111}<011>Pt;
- 模拟图像(图2c)与实验高度吻合,证实界面结构。
图2d–f:SnOx/Oct-Pt界面
- 3圈沉积(图2d):同样形成1–2原子层SnOx,界面氧原子清晰;
- Sn–Pt层间距2.3 Å,与Pt(111)面间距相当;
- 10圈沉积(图2e):SnOx增厚至3–5层,形成金红石相SnO₂,外延取向{100}<011>SnO₂//{111}<011>Pt;
- 晶格失配仅4.7%,导致高质量外延生长;
- 模拟图像(图2f)与实验完美匹配。
图2g:SbOx/Oct-Pt界面
- 完全非晶,无外延关系,与Pt(111)晶格失配高达30.6%。
图2h–i:EDX元素面扫
图2j:EELS谱
- InOx/Pt表面检测到In M边和O K边,确证氧化物存在。
图2k:晶格失配统计
- SnO₂(100)与Pt(111)失配最小(4.7%),In₂O₃(111)次之(21.1%),Sb₂O₃(111)最大(30.6%),与氧化物结晶性顺序一致。
图3:MOx/Pt界面的电位依赖动态结构与ORR活性
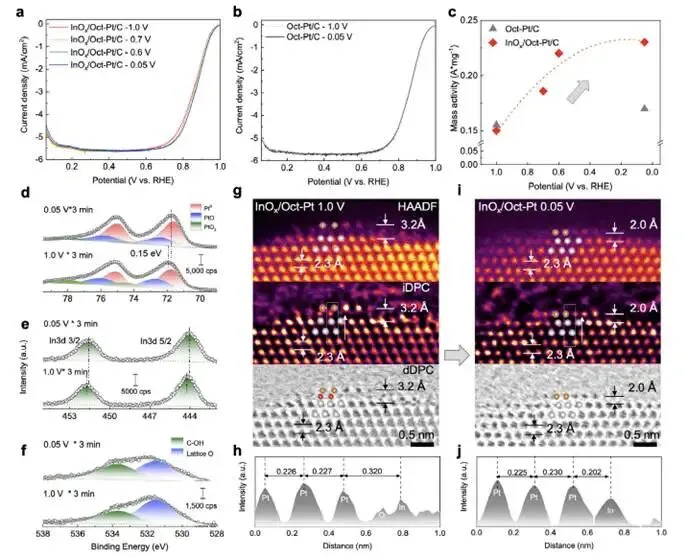
图3a–c:电位调控的ORR活性
- 将InOx/Oct-Pt/C电极在1.0、0.7、0.6、0.05 V各恒电位处理3 min后测试ORR极化曲线;
- 随处理电位降低,ORR活性逐渐升高,0.05 V处理后质量活性提升35%;
- 对比Oct-Pt/C(图3b),活性提升幅度远小于InOx/Pt,证明界面是关键。
图3d–f:XPS揭示电位诱导的化学态变化
- 0.05 V处理后(vs. 1.0 V):
- Pt 4f结合能负移,Pt⁰比例从53%升至68%(表S1);
- In 3d结合能负移0.2 eV;
- O 1s中晶格氧(530.6 eV)比例下降;
- 结论:低电位下InOx发生部分还原,氧空位增加。
图3g–j:STEM直接观测界面结构转变
- 1.0 V处理后(图3g–h):界面保持In–O–Pt结构,In–Pt间距3.2 Å;
- 0.05 V处理后(图3i–j):界面氧原子层消失,In–Pt间距缩短至2.02 Å,形成氧缺陷的Pt–In金属键界面;
- 动态“呼吸”模型确立:高电位(氧化态):Pt–O–M界面(富氧)→ 抑制Pt溶解 → 稳定性;低电位(还原态):Pt–M界面(缺氧)→ 电子转移 → 活性。
图S9–S10:SnOx、SbOx同样呈现电位依赖还原,但还原程度In > Sn > Sb,与电负性差顺序一致。
图4:从模型催化剂到实用Pt/C催化剂的拓展
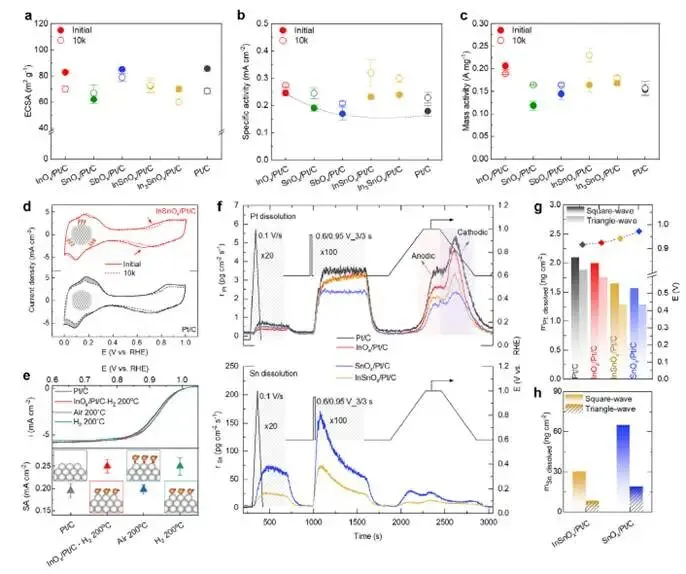
图4a–c:MOx/Pt/C(50 wt% Pt,~3 nm)的ORR性能
- 初始活性:InOx/Pt/C提升最显著(质量活性0.28 A mg⁻¹),SnOx/Pt/C次之(0.22),SbOx/Pt/C接近Pt/C(0.18);
- ADT后:
- InOx/Pt/C:ECSA下降17%,质量活性下降8%;
- SnOx/Pt/C:ECSA几乎不变,质量活性提升39%;
- SbOx/Pt/C:ECSA几乎不变,质量活性提升14%;
- 趋势:SnOx稳定效果最佳,InOx活性提升最佳。
图4d:复合氧化物InSnOx/Pt/C
- ECSA几乎不变(红曲线);
- 质量活性0.23 A mg⁻¹(ADT后),为Pt/C的1.5倍;
- 协同效应:Sn稳定+In活性。
图4e:热处理气氛诱导的界面可逆转变
- H₂还原(200°C)→ 形成Pt–In界面 → 活性↑;
- 空气氧化(200°C)→ 形成Pt–O–In界面 → 活性恢复至Pt/C水平;
- 二次H₂还原 → 活性再次↑;
- 完美可逆性,与电位诱导的“呼吸”行为一致。
图4f:operando ICP-MS监测Pt溶解
- 模拟方波ADT(0.6–1.0 V),实时检测电解液中Pt浓度;
- Pt溶解速率:Pt/C > InOx/Pt/C > InSnOx/Pt/C > SnOx/Pt/C;
- SnOx抑制Pt溶解效果最佳,与稳定性测试一致。
图5:MEA性能验证与PtCo催化剂拓展
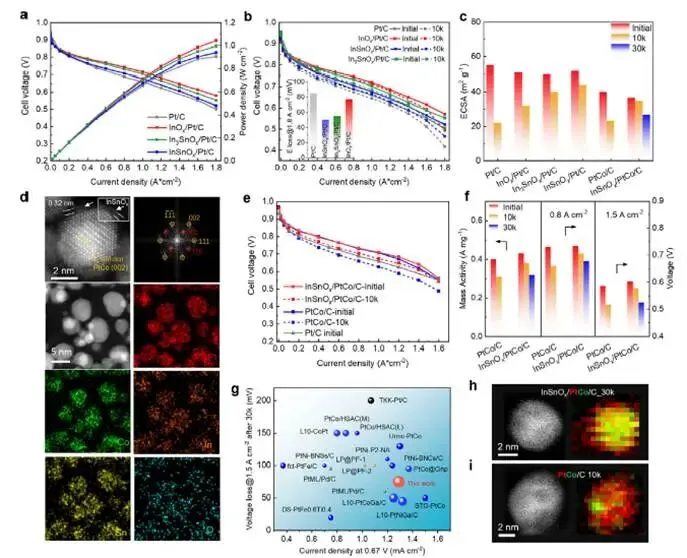
图5a–b:H₂-O₂ MEA极化曲线(80°C,100% RH,150 kPa)
- 初始性能:InOx/Pt/C、InSnOx/Pt/C、In₃SnOx/Pt/C均优于Pt/C;
- 10k方波ADT后:
- HCD区(>1.5 A cm⁻²)电压损失:InSnOx/Pt/C ≈ In₃SnOx/Pt/C(~50 mV)< InOx/Pt/C(77 mV)< Pt/C(85 mV);
- 与ECSA损失趋势一致(图5c)。
图5c:ECSA损失统计
- Pt/C损失45%,InOx/Pt/C损失30%,In₃SnOx/Pt/C损失20%,InSnOx/Pt/C损失18%;
- Sn含量越高,ECSA保持越好。
图5d:InSnOx/PtCo/C催化剂结构
- HAADF-STEM显示有序PtCo金属间化合物(FFT出现(001)超点阵衍射);
- EDX面扫显示In、Sn、Co、Pt均匀分布,InSnOx覆盖于PtCo表面。
图5e–f:H₂-air MEA性能
- 初始:InSnOx/PtCo/C性能优于PtCo/C和Pt/C;
- 10k ADT后:InSnOx/PtCo/C电压损失显著低于PtCo/C;
- ECSA损失:InSnOx/PtCo/C仅5%,PtCo/C达42%。
图5g:文献性能对比
- 30k ADT后,InSnOx/PtCo/C在0.67 V下电流密度达1.3 A cm⁻²(功率密度0.87 W cm⁻²),@1.5 A cm⁻²电压损失仅75 mV,位居已报道催化剂前列(表S6)。
图5h–i:Co溶出抑制
- InSnOx/PtCo/C的Co溶出量远低于PtCo/C;
- 归因于SnOx稳定的Pt壳层有效阻挡Co向外扩散。
五、总结与展望
本研究首次系统揭示了MOx/Pt(M = In, Sn, Sb)界面在燃料电池ORR电位区间内的动态“呼吸”行为,并成功利用这一行为在原子尺度上解耦了催化剂的活性与稳定性。
核心贡献:
- 机制层面:建立电位-界面构型-催化性能的直接关联,阐明低电位Pt–M界面→活性、高电位Pt–O–M界面→稳定性的原子机制;
- 方法层面:提出“活性剂+稳定剂”复合界面策略,实现In(活性)与Sn(稳定)的协同优化;
- 性能层面:将策略成功拓展至实用高载量Pt/C和PtCo金属间化合物催化剂,在MEA级别验证了其优异的高电流密度性能与超长耐久性;
- 范式意义:“动态界面”概念的提出,打破了催化剂设计中活性与稳定性“非此即彼”的传统认知,为智能催化剂的发展开辟了新方向。
未来方向:
- 界面组分精准筛选:结合理论计算与高通量实验,探索更多p区金属(Ga、Ge、Bi等)及复合氧化物体系;
- 动态界面的工况谱学:发展operando XAFS、environmental TEM等技术,在真实MEA运行条件下实时追踪界面演化;
- 抗反极/抗杂质拓展:研究动态界面在启动/停机、空气杂质(SOx、NOx)等极端工况下的行为;
- 跨反应拓展:将“动态界面呼吸”概念推广至电解水、CO₂还原、氮还原等其他电催化体系。
六、通讯作者简介
干林,清华大学深圳国际研究生院材料研究院长聘副教授,博士生导师。主要从事高分辨电子显微学与能源材料研究。重点发展高时空与能量分辨的(原位)电子显微成像与谱学方法,在原子尺度上开展电化学能源材料的晶格、电荷、自旋和轨道等序参量的实验测量及其与性能的关联性研究,揭示能源材料在原位/工况条件下的表面和固液电化学界面结构及其动态变化,发展高性能和高稳定性的电化学能源材料与器件。迄今在Science、Nature Mater、Nature Commun、Adv Mater、Angew Chem Int Ed等期刊上发表论文100余篇,引用8000余次。获得国家自然科学基金委优秀青年基金、面上项目、广东省重点研发领域课题、广东省杰出青年科学基金和深圳市高等学校稳定支持计划等项目资助。获国际电子显微学会联合会青年科学家奖(IFSM Young Scientist Award 2014),深圳市国家级领军人才。
Marc Ledendecker,慕尼黑工业大学可持续能源材料课题组组长、青年教授。研究方向为电催化剂稳定性、电解水、燃料电池。在Angew. Chem. Int. Ed.、Adv. Mater.、Energy Environ. Sci.等期刊发表论文40余篇。获德国联邦教育与研究部(BMBF)NanoMatFutur项目资助。
声明:文章内容仅代表本人解读观点,不代表原作者及期刊立场。如有侵权,请联系后台删除。感谢支持,欢迎批评指正!
欢迎各位老师、同学在公众号上投稿分享课题组研究成果,不限期刊和发表时间!后台联系即可。
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
