跻身最佳非贵金属催化剂之列!深圳大学&哈工大&燕大最新AM!
- 2026-05-31 09:50:41
跻身最佳非贵金属催化剂之列!深圳大学&哈工大&燕大最新AM!
【做计算 找华算】华算科技新年献礼,经费预存高至30%增值,更有8500+返利直接送!限时福利,快冲! 电催化性能由活性位点周围的即时微环境——特别是稳定反应中间体的氢键网络——所决定。虽然水相电解液中的阳离子效应可用于调节该网络,但在质子交换膜燃料电池(PEMFC)中,质子是唯一阳离子,这一有力杠杆缺失。 
在本文中,作者提出了一种适用于单原子催化剂的“固定化分子扰动”通用策略,将调节功能从电解液转移到催化剂的第二配位层。以Fe-N-C上的氧还原反应(ORR)为模型,作者证明近端P-O基团可作为空间位阻和氢键扰动剂。该工程化微环境选择性削弱关键*OH中间体的溶剂化壳层(光谱和计算证实),从而促进速率决定步骤——*OH脱附。 这一调控赋予催化剂卓越性能:在0.5 M H2SO4中半波电位达0.861 V,H2/O2PEMFC峰值功率密度达1024 mW cm-2;同时在0.65 V下运行253 h后电流保持72%,跻身文献报道的最佳非贵金属催化剂之列。本工作将范式从单纯优化活性中心转向有目的地设计局域微环境,实现器件相关环境下电催化加速。 质子交换膜燃料电池(PEMFC)被普遍视为未来可持续能源格局的关键技术之一。然而,其大规模商业化受到阴极氧还原反应(ORR)动力学迟缓和昂贵铂基催化剂严重依赖的制约。为应对这一挑战,非贵金属–氮–碳(M-N-C)单原子催化剂被公认为有前途的替代方案。近年来,通过电子结构调控和缺陷工程取得的显著进展,使其性能大幅提升,成为替代传统Pt/C的可行候选。然而,目前的研究工作仍主要聚焦于提升这类催化剂的本征活性,却在很大程度上忽视了工作条件下受限在双电层内的界面水微环境所发挥的关键作用。这一疏漏尤为关键,因为M-N-C催化剂的活性位和作用机制与Pt/C根本不同:M-N-C以孤立金属原子为活性位,实现与反应分子的“点对点”相互作用;而Pt/C则拥有延展晶面,进行“面对点”相互作用。M-N-C活性位高度局域的特性使其性能对电极–电解液界面局部水微环境异常敏感。 先进原位表征和理论方法的不断发展,日益凸显催化剂–电解液界面处界面水微环境的关键作用。这一独特环境深刻影响质子转移动力学、调变氧中间体结合强度,甚至可决定反应路径。目前,调控这一无序氢键水网络的研究主要依赖外部策略,如电解液添加剂、表面修饰剂和离子液体等。尽管这些方法在一定程度上改善了ORR动力学,但存在诱导副反应、环境适应性差、对界面水结构控制缺乏精度等固有缺陷。更关键的是,外来分子物种的引入增加了微环境复杂性,缺乏靶向操控不仅带来性能不确定性,更遮蔽了*OH解离等关键步骤中催化剂–微环境协同作用,掩盖了基本的构效关系,阻碍了对ORR机理的深入理解。因此,通过催化剂本征工程实现对局部水微环境的精准控制,对于推进ORR机理基础认知和高性能催化剂靶向设计至关重要。 基于上述分析,作者提出在Fe单原子第二配位层引入P–O基团,通过精准调控界面水氢键网络以提升ORR性能。作者首先从理论上论证该结构的可行性及其对催化动力学整体提升的贡献。具体而言,通过从头算分子动力学(AIMD)模拟与原位光谱分析相结合,作者发现P–O基团与界面水结合形成的P–O···H3O+(H2O)x空间位阻效应,显著削弱了*OH中间体与周围水分子之间的氢键相互作用;该效应大幅降低*OH脱附能垒,促进其快速解离。随后,利用植酸蚀刻策略,作者在碳基底上成功构筑了PO/FeN4构型催化剂(记为PO/Fe-N-C),并通过多种结构表征技术进行了系统验证。 得益于这一独特结构,PO/Fe-N-C催化剂展现出优异的ORR电催化性能:半波电位达0.861V,10000次循环后衰减仅7 mV。应用于PEMFC时,在H2/O2条件下最大功率密度达1.024 W cm-2,10000次耐久测试后性能保持80%;在H2/air条件下,最大功率密度为0.501 W cm-2,0.65 V运行253 h后电流密度保持72%。凭借高膜电极活性和稳定性,PO/Fe-N-C催化剂跻身迄今报道最佳M-N-C催化剂之列。 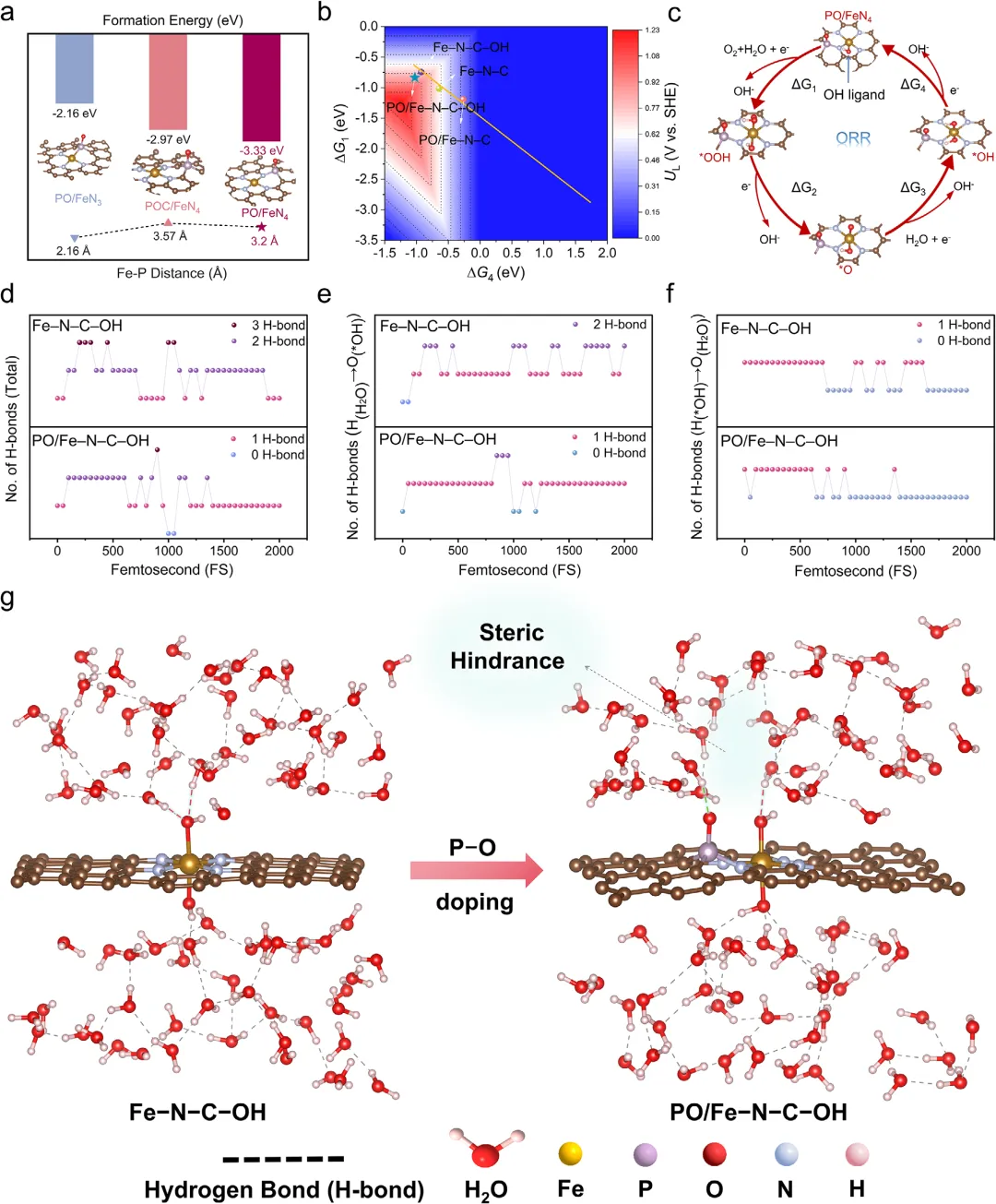
图1:理论计算揭示第二配位层P─O基团调控*OH脱附机理。a)三种P-O位置模型形成能:第二壳层PO/FeN4最负(−3.33 eV),Fe-P间距≈3.20 Å。b)二维火山图:PO/FeN4-OH极限电位UL最高(0.84 eV),RDS由*OOH形成转为*OH脱附。c)完整ORR路径:PO/FeN4-OH的*OH脱放能垒最低。d-f)AIMD氢键统计:PO/FeN4=OH中*OH与界面水总氢键数、O···H与H···O氢键均显著减少。g)界面快照:P-O基团产生空间位阻,削弱*OH溶剂化壳,促进快速脱附。 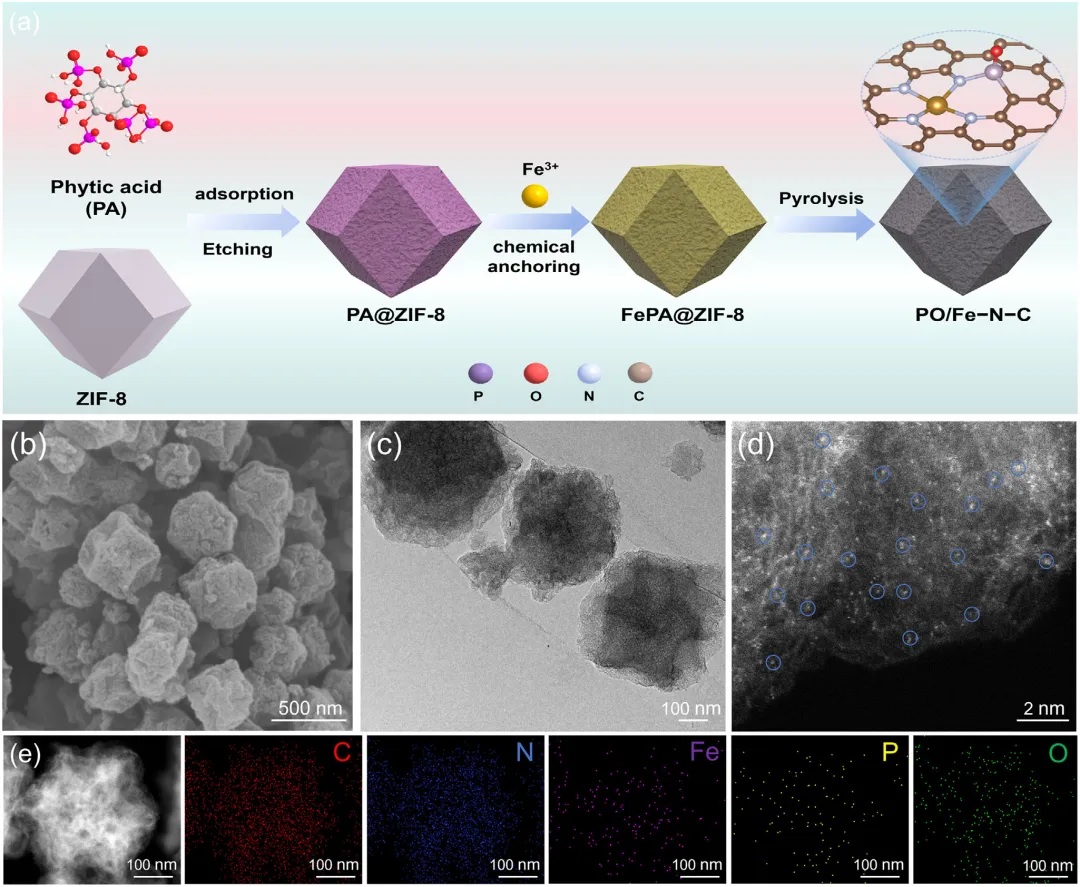
图2:PO/Fe-N-C合成与形貌结构。a)合成路线:植酸蚀刻ZIF-8→Fe3+锚定→1050 °C碳化,获得PO/Fe-N-C。b)SEM:保持菱形十二面体,表面皱缩多孔。c)TEM:无Fe纳米颗粒,骨架完整。d)AC-HAADF-STEM:原子级分散亮斑点为Fe单原子。e)EDS元素分布:C、N、Fe、P、O均匀共分布。 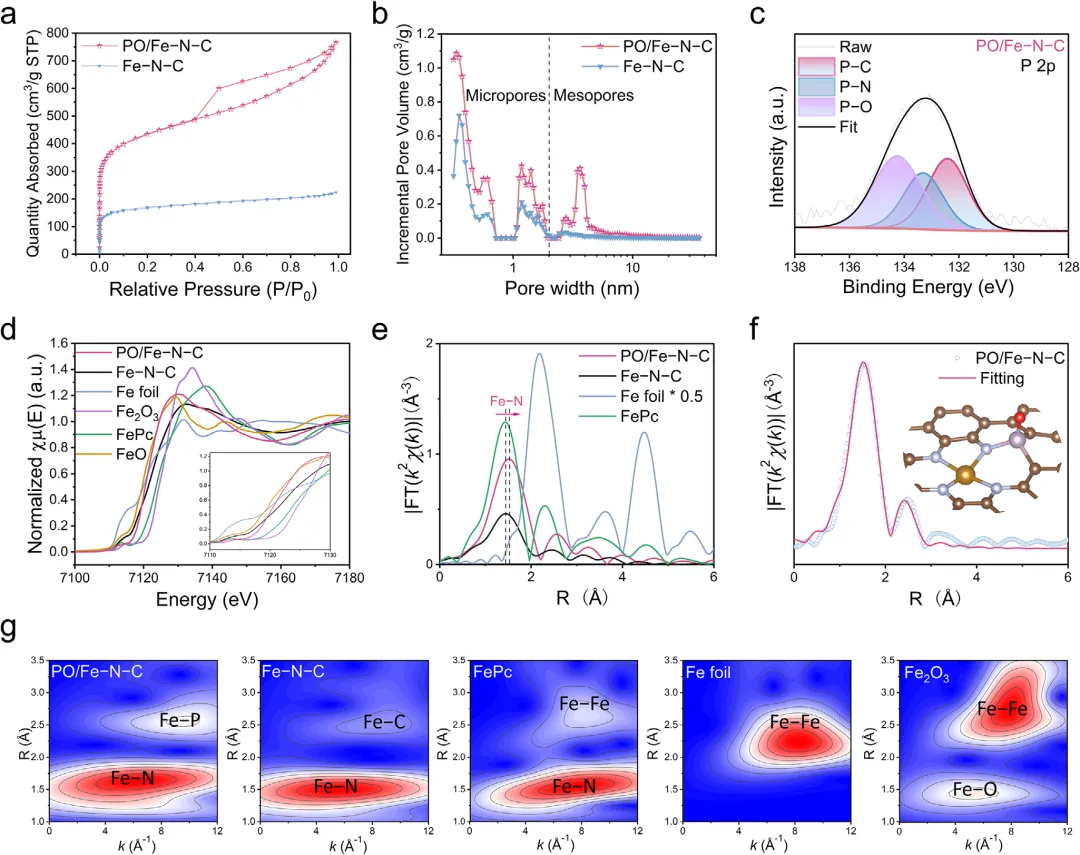
图3:孔结构、元素价态与配位环境。a-b)BET:PO/Fe-N-C比表面积1590 m2 g-1(≈2.5×Fe-N-C),介孔/微孔体积均大幅提升。c)P 2p XPS:132.3eV(P-C)、133.2eV(P-N)、134.3eV(P-O)共存。d)FeK-edge XANES:白线强度负移,Fe价态更接近+2。e)FT-EXAFS:仅Fe-N/O散射峰,无Fe-Fe,证实原子分散;PO/Fe-N-C Fe-N键长略增,表明P诱导局部畸变。f)R空间拟合:Fe-N CN≈3.96,Fe-P CN≈1.21,P位于第二壳层。g)小波变换:强度峰位与FePc接近,区别于Fe箔/氧化物,进一步确认FeN4构型。 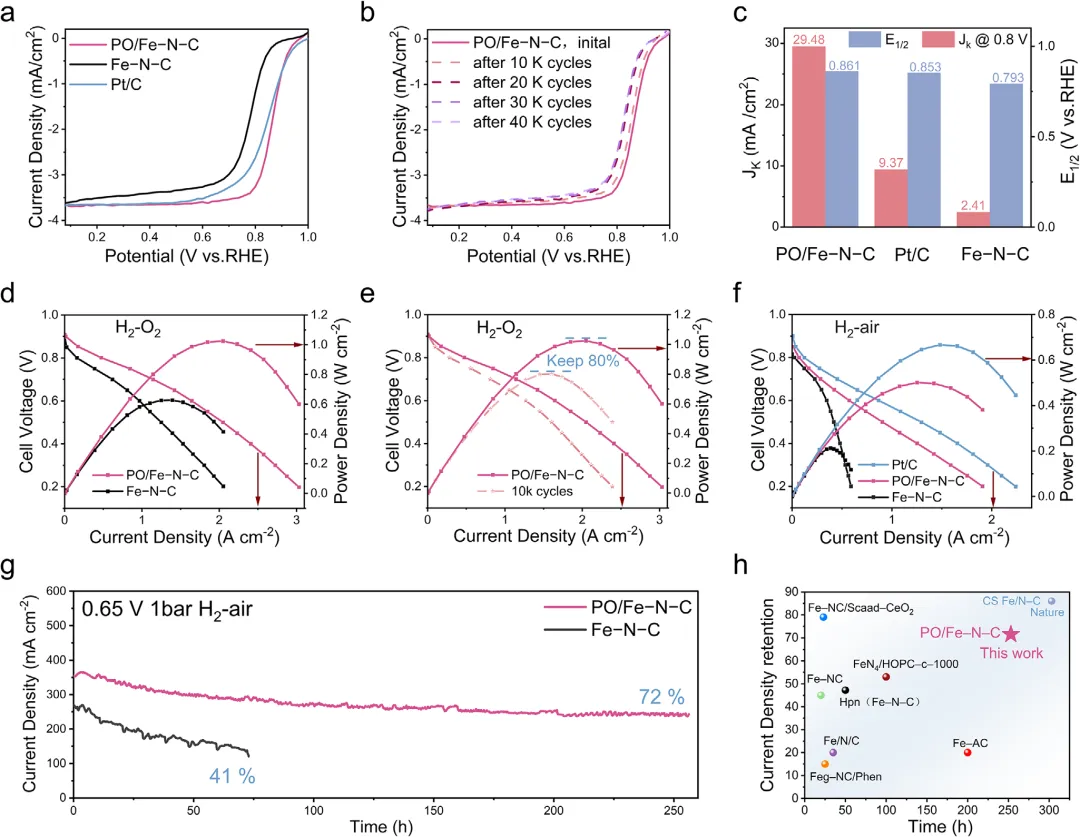
图4:酸性ORR与PEMFC性能。a)RRDE极化曲线:PO/Fe─N─CE1/2=0.861V,比Fe─N─C高68mV,优于商业Pt/C。b)耐久性:10000/40000圈后E1/2仅下降7/25mV。c)动力学比较:Jk(0.8V)达29.5mA cm-2,质量活性1519mA mg-1,TOF0.88es-1,均优于对照。d)H2/O2PEMFC:峰值功率密度1024mW cm-2(Fe─N─C 627mW cm-2)。e)AST 10000圈后PO/Fe─N─C保留80%Pmax,Fe─N─C仅70%。f)H2/air PEMFC:Pmax=501mW cm-2,远高于Fe─N─C 212mW cm-2。g-h)恒压0.65V耐久:PO/Fe─N─C 253h后保持72%电流,Fe─N─C 71h后仅41%;电荷/传质阻抗均更低。 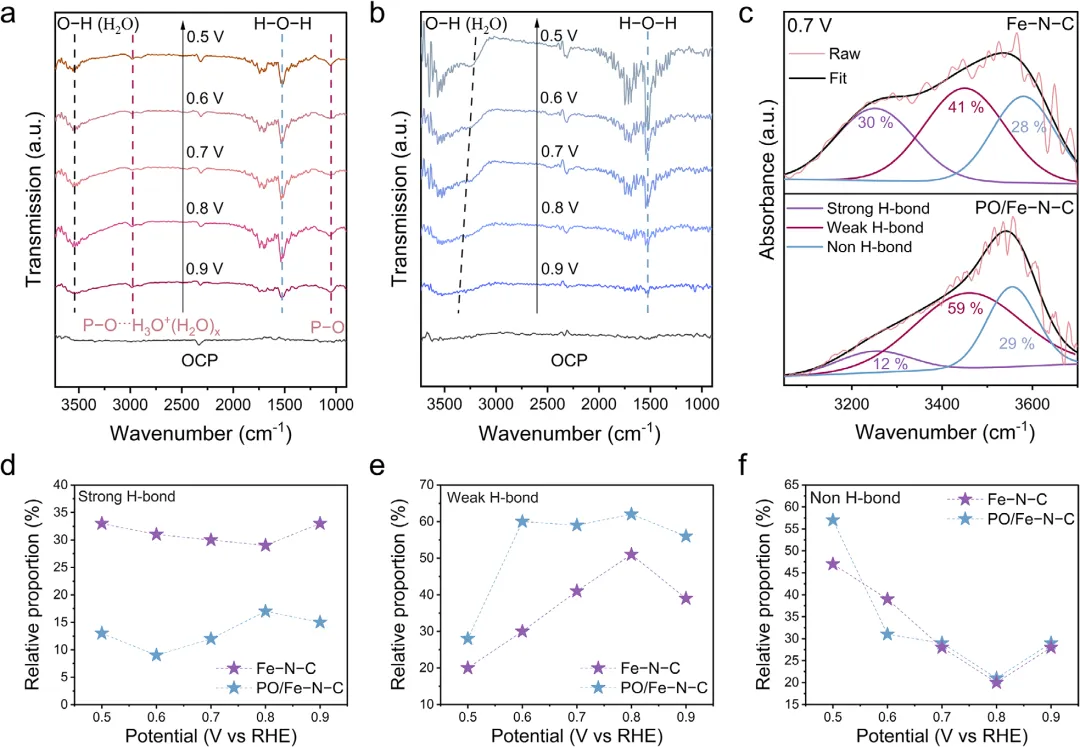
图5:原位FT-IR验证界面水氢键削弱。a-b)原位FT-IR:PO/Fe─N─C出现P─O···H3O+(H2O)x特征峰(~1050与2890–2980cm-1),随电位同步变化。c)O–H伸缩区高斯分峰:4-HB⋅H2O(强氢键)占比仅~13%,Fe─N─C达~33%;2-HB⋅H2O(弱氢键)比例显著升高。d-f)水物种分布:PO/Fe─N─C界面强氢键水大幅减少,孤立水分子增加,与AIMD结论一致,证实P─O位阻削弱*OH溶剂化。 综上,作者提出“固定分子扰动”策略,在Fe-N-C单原子催化剂第二配位球中精准引入P-O基团,通过空间位阻与氢键协同作用削弱*OH中间体与界面水的氢键网络,降低*OH脱附能垒,从而突破酸性氧还原反应(ORR)动力学瓶颈。 本工作首次将“界面水氢键工程”内嵌于催化剂本征结构,摆脱了对电解液添加剂的依赖,为质子交换膜燃料电池及其它质子介质电催化过程提供了可扩展、高活性、长寿命的非贵金属单原子催化剂设计范式,有望大幅降低燃料电池成本并推动其在交通与分布式能源领域的商业化应用。 Breaking the Intermediate Solvation Shell on Single-Atom Catalysts With a Proximal Group Perturber for Enhanced Oxygen Reduction. Adv. Mater., 2026. https://doi.org/10.1002/adma.202523627. 
👉 点击阅读原文,立即下单!
2026年1月26日,深圳大学隋旭磊、郑勇平、燕山大学邵光杰、哈尔滨工业大学王振波在国际知名期刊Advanced Materials发表题为《Breaking the Intermediate Solvation Shell on Single-Atom Catalysts With a Proximal Group Perturber for Enhanced Oxygen Reduction》的研究论文,Zhaoyang Han、Ruihui Gan、Tao Gong为论文共同第一作者,隋旭磊、郑勇平、邵光杰、王振波为论文共同通讯作者。
本文来自网友投稿或网络内容,如有侵犯您的权益请联系我们删除,联系邮箱:wyl860211@qq.com 。
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 东莞市厚街镇接送病人长途出院车:收费标准、长途护送、转院出院、跨省护送服务、长途转运
- 广州中医药大学顺德医院,2026年人才招聘启事(含护理22名)
- 唐忠阳、夏昆赴广州访企拓岗 推动校企合作和“校友回湘”工作
- 企业动态 | 广州安居集团推动新开工保障房项目4个,共计7425套
- 东莞市石龙镇接送病人长途出院车:长途跨省护送、接送病人、非急救120收费标准、重症转诊
- 东莞市长安镇接送病人长途出院车:护送服务-出院转院-高铁航空转运-120接送病人(重症转诊)
- 10570广州医科大学25年招生专业&录取分数
- 【深圳保安招聘】1月29日
- 报名开启 | 深圳青少年公开赛·2026首战观澜湖
- 东莞市石碣镇接送病人长途出院车:正规非急救120护送、跨省长途、预约平台、护送服务
