铁死亡是一种由脂质过氧化失控驱动的非凋亡性细胞死亡形式,在肿瘤治疗中展现出巨大潜力。通过药理学诱导铁死亡已成为一种有前景的抗癌策略,但肿瘤细胞对铁死亡产生的耐药性构成了重大挑战。脂质代谢受复杂信号网络调控,深刻影响铁死亡的敏感性和耐受性。含多不饱和脂肪酸的磷脂(PUFA-PLs)特别容易发生过氧化反应,其合成需要长链酰基辅酶A合成酶4(ACSL4),并通过溶血磷脂酰胆碱酰基转移酶3(LPCAT3)运输到细胞膜。过氧化物酶来源的PUFA缩醛磷脂和脂噬介导的脂质降解产物也是脂质过氧化和铁死亡的底物。
在此背景下,谷胱甘肽过氧化物酶4(GPX4)在阻止铁死亡中扮演关键角色。GPX4能将膜磷脂氢过氧化物(PL-OOH)转化为相应的醇类(PL-OH),从而防止含氧脂质氢过氧基中间体的毒性裂解。然而,GPX4催化的PL-OH脂肪酸链中亲水基团的极性仍可能导致细胞膜损伤。磷脂酶A2(PLA2)家族酶能在sn-2位置水解膜磷脂,释放溶血磷脂和游离脂肪酸,从而消除氧化还原脂质死亡信号。过氧化还原蛋白6(PRDX6)作为一种钙非依赖性细胞内PLA2发挥功能,其活性受pH水平调控。在细胞质pH下,PRDX6主要结合并水解氧化磷脂。尽管已描述了几种GPX4非依赖性通路,GPX4在各种模型中抑制铁死亡仍发挥核心作用。
近期,广州医科大学基础医学院刘金保/中山医学院李隽在Molecular cell期刊联合发布题为Targeting PRDX6-dependent localization and function of GPX4 enhances ferroptosis-mediated tumor suppression的文章,描述了铁死亡防御的PRDX6依赖机制,为靶向癌症治疗提供了新的视角。
1) 脂质组学分析揭示过氧化应激诱导溶血磷脂积累:研究者通过质谱分析发现,经erastin或BSO处理的细胞中溶血磷脂(特别是LPC和LPE)水平显著升高。磷脂组学进一步显示,非氧化型LPC(16:1、18:0、18:1)和LPE(16:0、18:0、18:1)是主要升高成分。氧化型多不饱和脂肪酸如15-HpETE显著增加,同时检测到氢过氧基-PE和氢过氧基-PC物种上调。这些结果表明过氧化应激通过细胞内氢过氧化磷脂水解促进溶血磷脂产生。
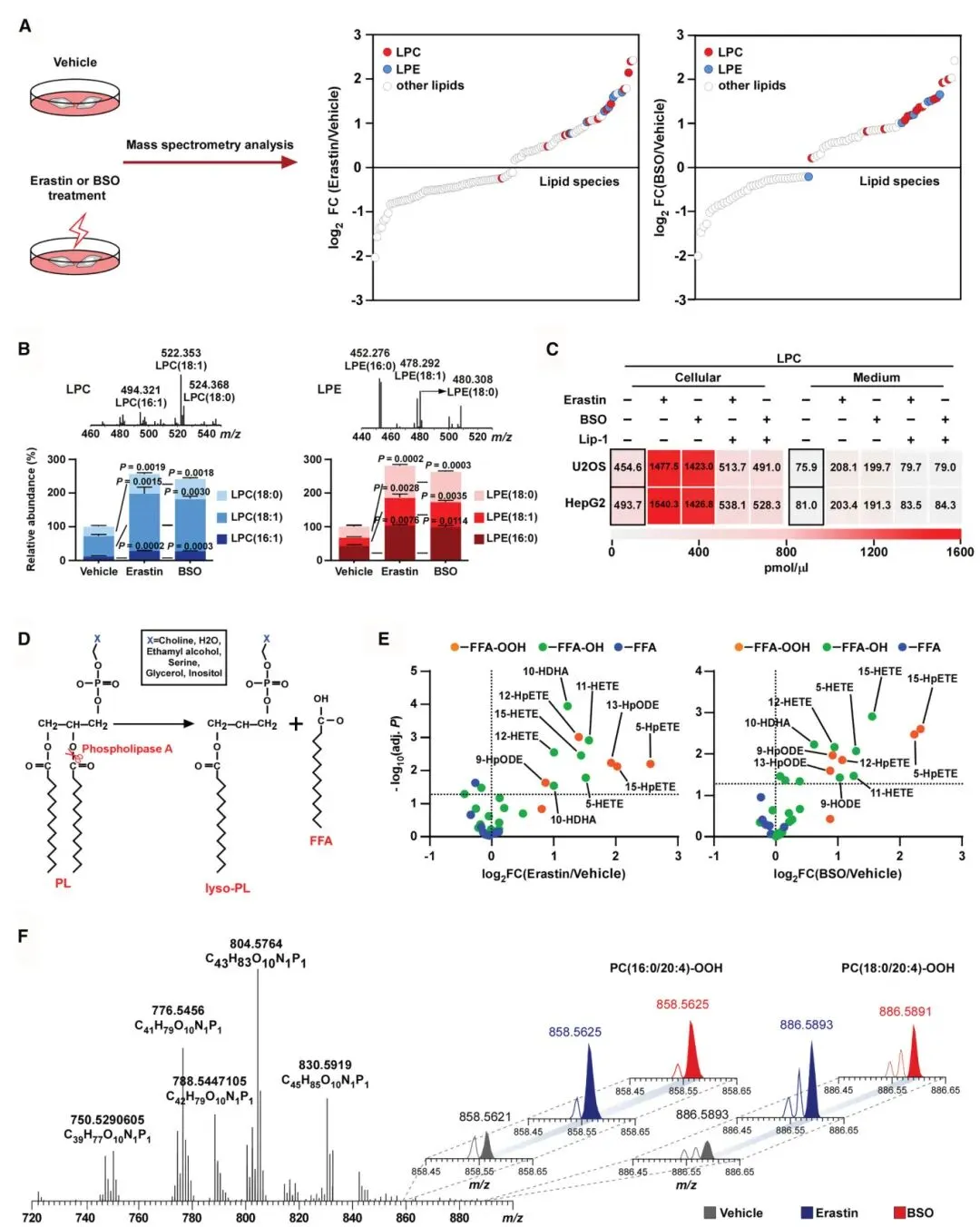
图1 过氧化应激诱导过氧化物PLs水解成溶血磷脂
2) PRDX6介导过氧化磷脂水解并抑制脂质过氧化:通过RNAi筛选35个PLA2家族成员,研究确定PRDX6对LPC积累具有最强抑制作用。PRDX6敲低使erastin或BSO诱导的PLA2活性降至约30%,证明PRDX6在过氧化应激期间对PLA2介导的磷脂水解贡献显著。PRDX6缺失导致氢过氧基-PE和氢过氧基-PC积累增加,Liperfluo染色显示脂质过氧化信号增强。重新引入PRDX6能减弱脂质过氧化,确认了PRDX6在防止脂质过氧化中的作用。
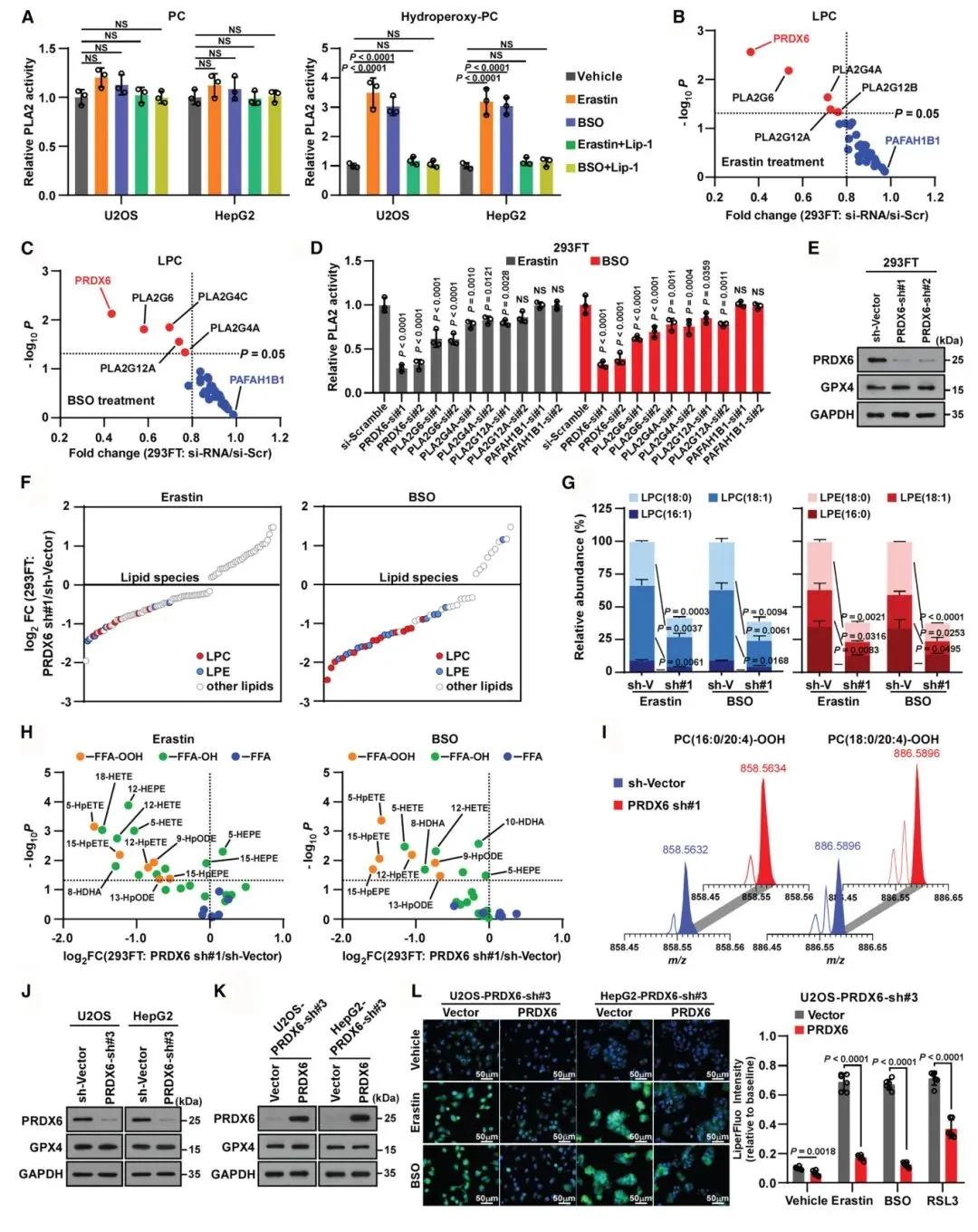
图2 PRDX6减轻过氧化应激诱导的过氧化物PLs水解,防止脂质过氧化
3) 过氧化应激通过二硫键诱导PRDX6-GPX4复合物形成:共免疫沉淀实验显示PRDX6与GPX4相互作用,该相互作用在erastin、BSO或RSL3处理后增强。免疫荧光显示PRDX6和GPX4在过氧化应激后共定位于细胞膜。非还原性PAGE分析显示约42 kDa条带对应GPX4-PRDX6异二聚体,该条带仅在非还原条件下出现。定点突变和共免疫沉淀揭示PRDX6的C47残基与GPX4形成分子间二硫键,C47S突变破坏了该相互作用,而C91S突变保留了野生型特征。
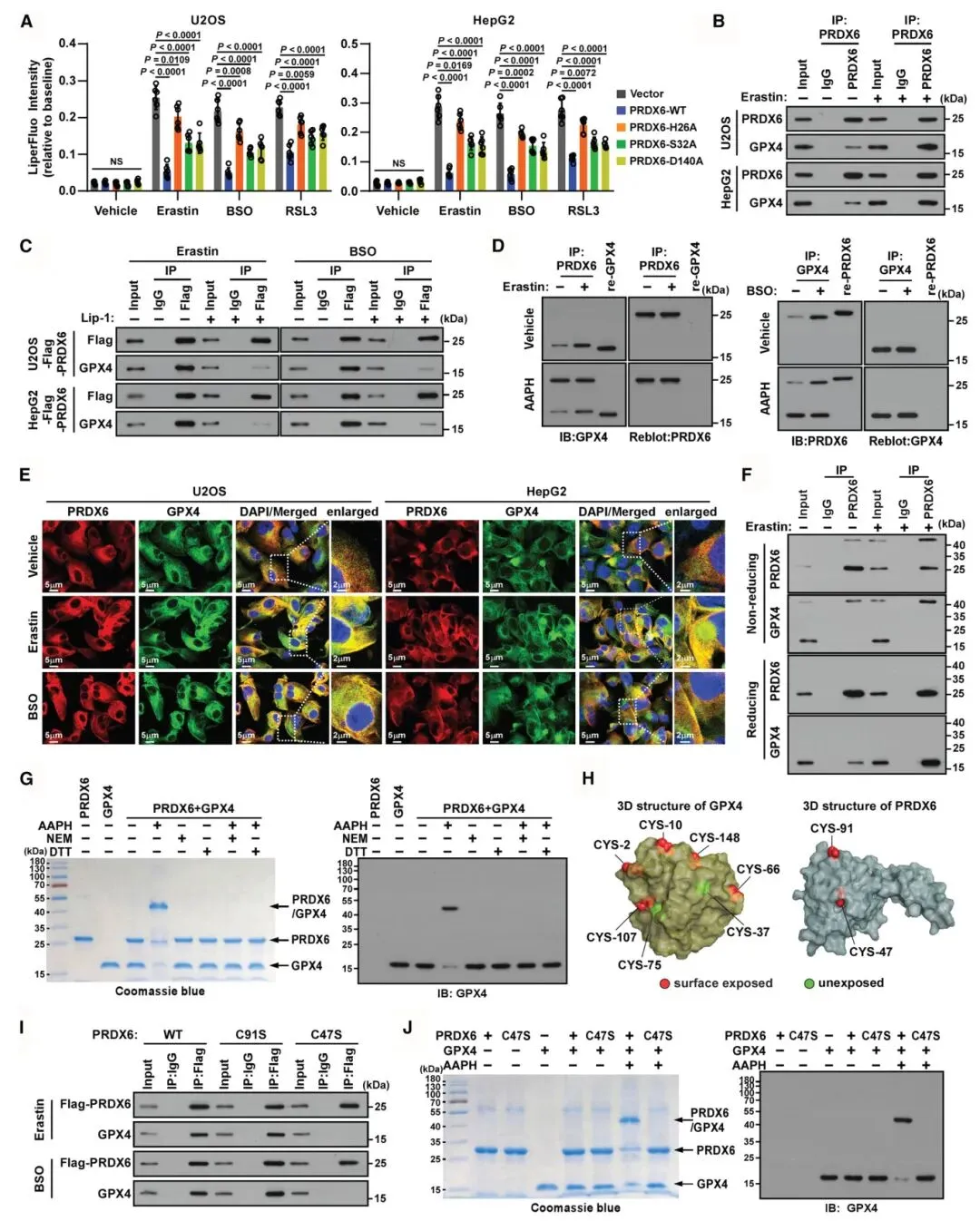
图3 过氧化应激通过二硫桥诱导PRDX6/GPX4复合物形成
4) PRDX6-GPX4复合物驱动GPX4膜转运:膜分级和免疫荧光实验显示,erastin或BSO处理促使PRDX6和GPX4转运至细胞膜。PRDX6敲低阻止了过氧化应激诱导的GPX4膜转运,而GPX4缺失不影响PRDX6的膜转运,表明PRDX6招募GPX4至质膜。重新引入PRDX6能剂量依赖性恢复膜组分中GPX4水平,但C47S突变显著降低GPX4的膜转运。微滤实验显示PRDX6-GPX4异二聚体与氧化的PC/PE中性脂质体结合,而GPX4单独不能结合,证明PRDX6促进GPX4与电中性磷脂结合。
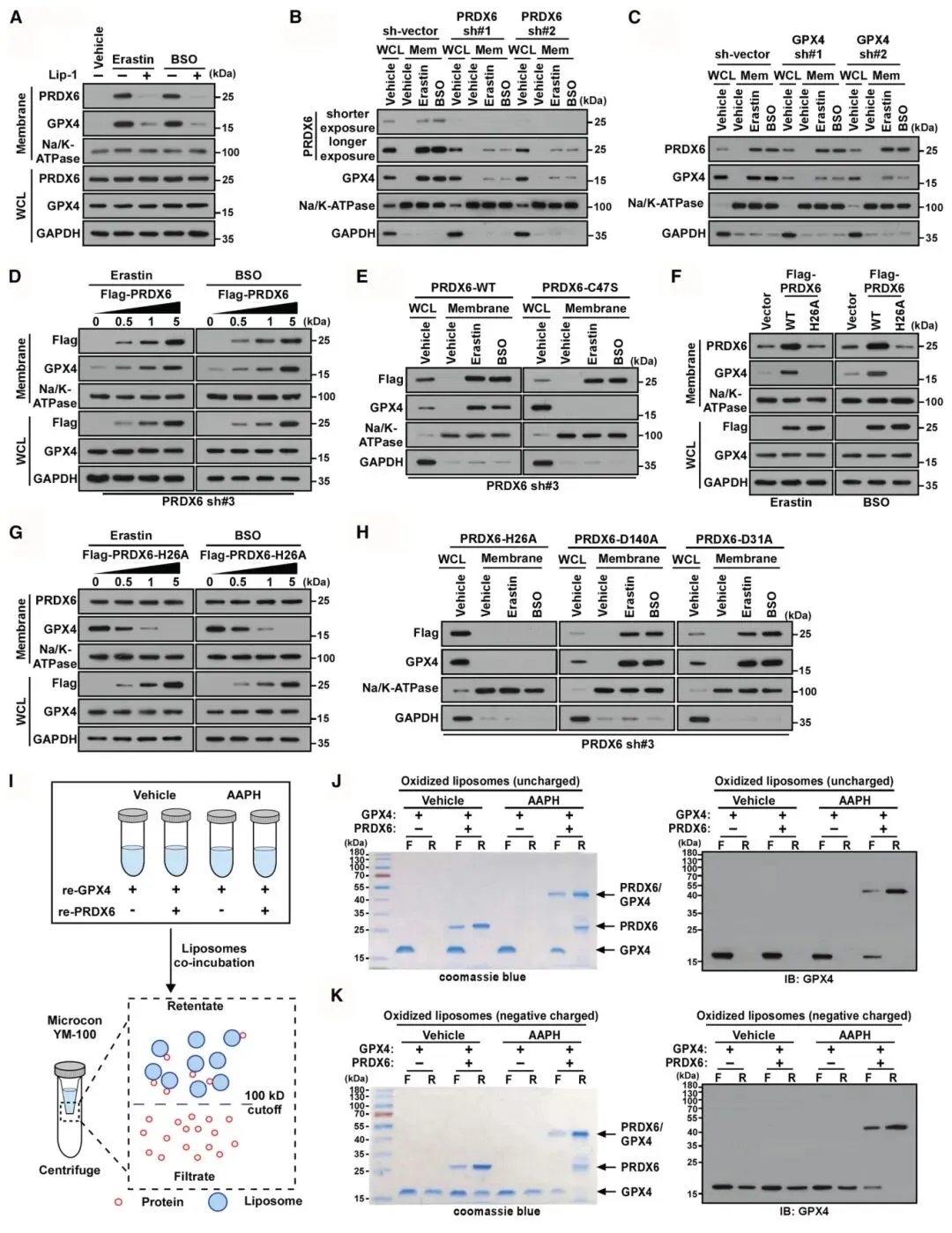
图4 PRDX6/GPX4复合物促进GPX4与羟基- pe和PC的结合和PHGPX活性
5) PRDX6-GPX4复合物通过双重机制修复过氧化膜:体外实验显示,重组PRDX6与1-SA-2-15-HpETE-PE共培养产生溶血磷脂和15-HpETE;加入纯化GPX4进一步产生溶血磷脂和15-HETE,同时减少15-HpETE。通过抗GPX4抗体中和GPX4后加入PRDX6,主要产生来自GPX4还原的PL-OH的15-HETE。PRDX6能水解PL-OH和PL-OOH,证明PRDX6-GPX4复合物作为消除磷脂氢过氧化物双重机制的平台。在PRDX6敲低细胞中,GPX4过表达仅部分减少脂质过氧化和膜损伤,强调了PRDX6在GPX4依赖和非依赖性铁死亡通路中的特异性作用。
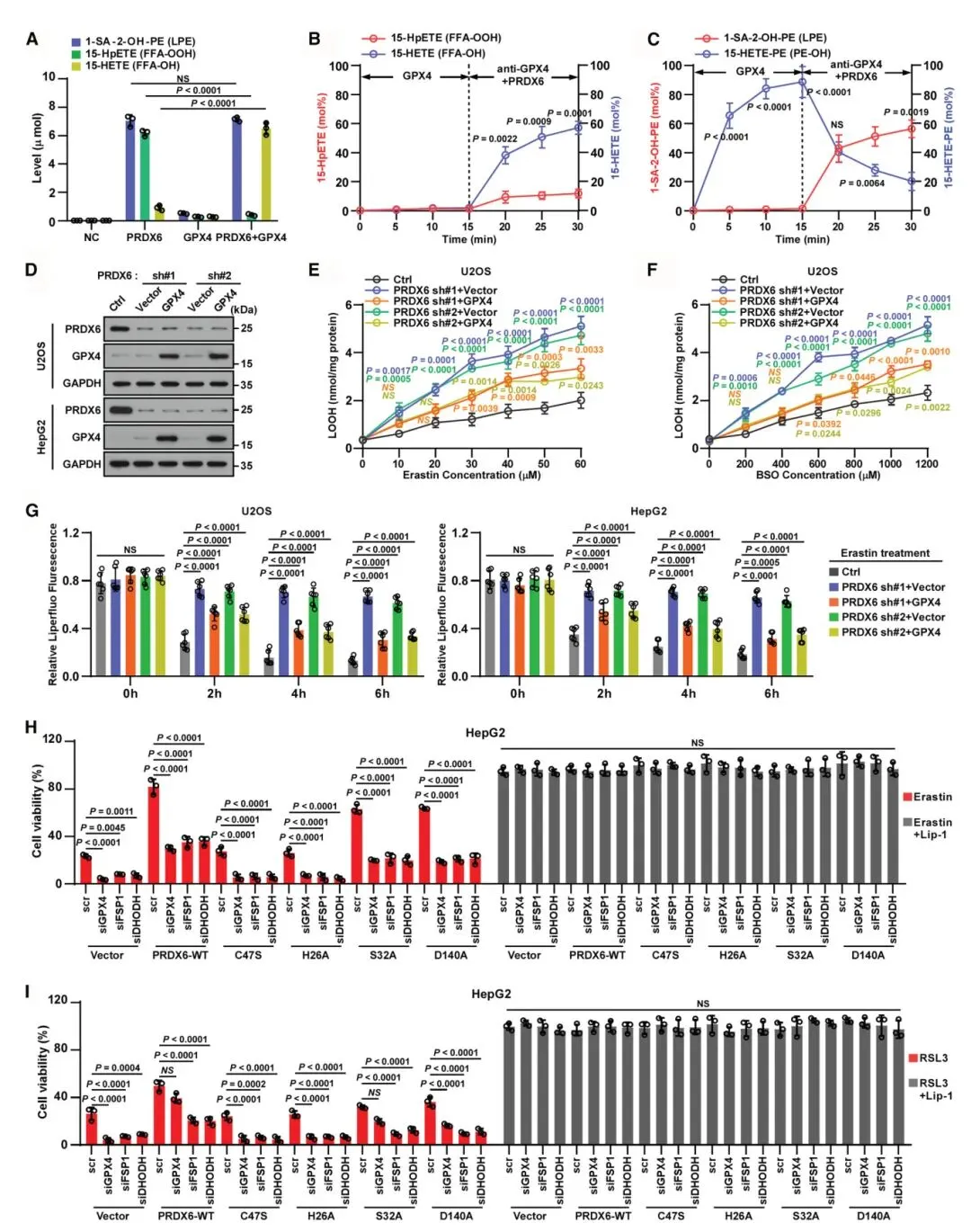
图5 PRDX6/GPX4复合物通过双重机制促进过氧化物膜的修复
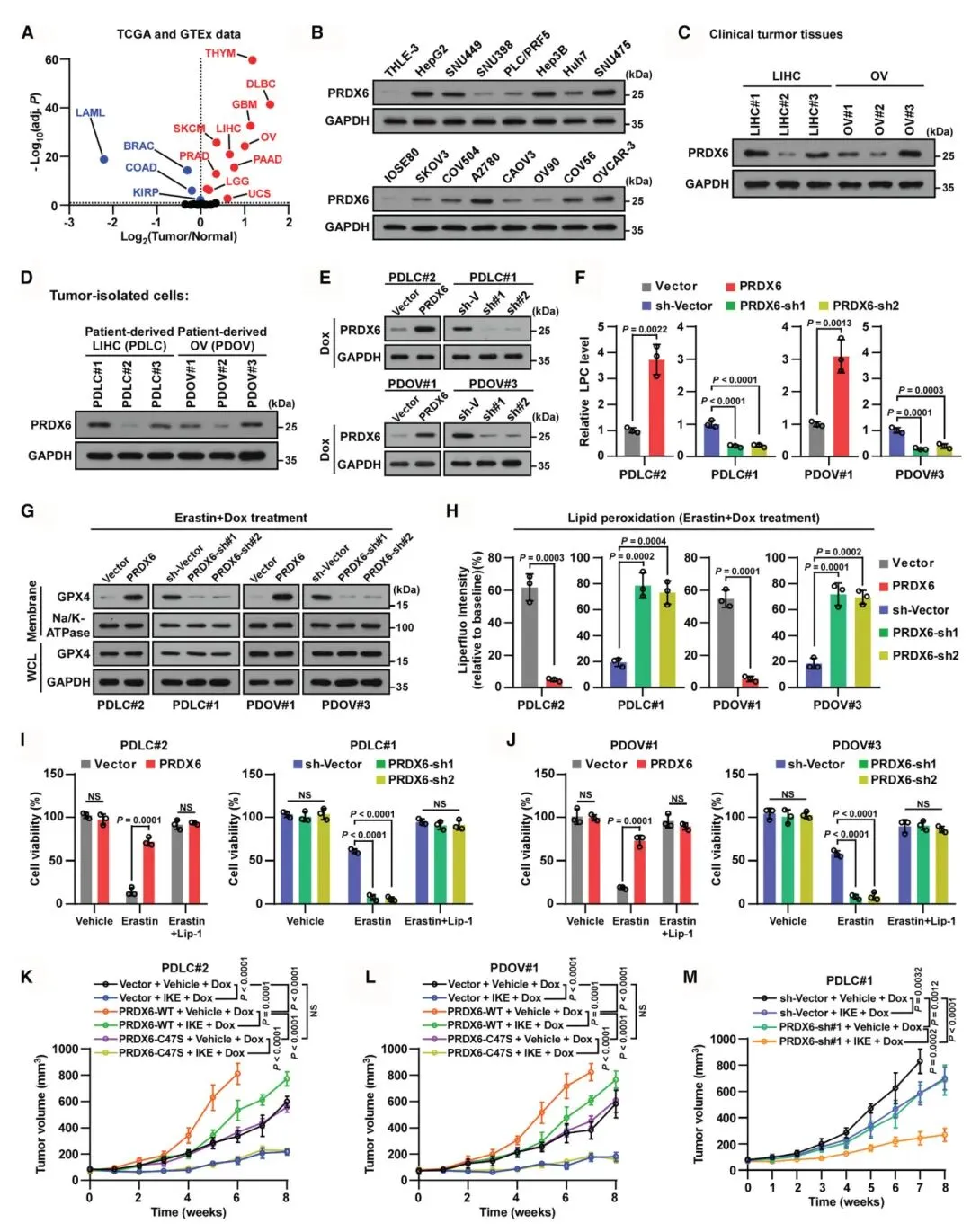
6) PRDX6上调通过抑制脂质过氧化促进肿瘤发展:多种癌症患者队列分析显示PRDX6高表达与较短的无进展生存期相关。在肝癌和卵巢癌患者来源细胞中,PRDX6过表达增加细胞LPC水平、膜GPX4表达,减少脂质过氧化并增强erastin处理的细胞活力。在小鼠异种移植模型中,PRDX6野生型和磷脂酶功能正常的突变体(S32A、D140A)过表达加速了IKE治疗下的肿瘤生长,但C47S和H26A突变体无此效应。患者来源异种移植(PDX)模型显示,AAV介导的PRDX6敲低联合IKE治疗在高PRDX6表达肿瘤中显著抑制肿瘤生长,提高4-HNE和MDA水平但不影响凋亡标志物。
图6 PRDX6过表达通过抑制脂质过氧化促进肿瘤发展