电催化硝酸盐还原反应(NO₃RR)利用可再生电力转化废水中的硝酸盐污染物,代表了一种可持续氨合成途径,但其在中性介质中的效率受到迟缓动力学和析氢反应(HER)激烈竞争的严重限制。
2026年03月01日,深圳大学何传新、张雪团队在Angewandte Chemie International Edition期刊发表题为“Self-Sustaining Dynamic Alkaline Microenvironment-Mediated Efficient Nitrate Electroreduction to Ammonia on MnFeOx in Neutral Electrolyte”的研究论文,团队成员贾欣美为论文第一作者,何传新、张雪为论文共同通讯作者。
第一作者:贾欣美
通讯作者:何传新、张雪
通讯单位:深圳大学
论文DOI:10.1002/anie.4598330
该研究介绍了一种由MnFe双位点氧化物实现的“自维持碱性局域微环境”策略,该氧化物同时作为结构支架和催化介质,其中FeOₓ中无活性FeOh位点被Mn选择性取代,而活性FeTd位点得以保留。一维MnFeOₓ中的Fe位点活化NO₃⁻并动态捕获OH⁻以形成FeOOH,在电极-电解液界面活性位点周围建立起局域碱性微环境,有效抑制了HER。同时,Mn位点稳定高价态Fe物种,并持续将界面H₂O解离为OH⁻和H*,确保了碱性微环境的稳健维持。所得一维MnFeOₓ催化剂在中性介质中实现了95.9%的NH₃法拉第效率(12.3 mg h⁻¹cm⁻²),并能稳定运行超过20小时而无性能衰减。该研究将局域pH调控从外部干预推进到智能自调节,为自适应电催化剂设计以及超越NO₃RR的界面微环境调控提供了新见解。
全球氮循环是地球生物圈的基石,它控制着养分的可用性,并维持着陆地和水生生态系统的初级生产力。然而,来自农业径流、工业废水和城市污水的过量硝酸盐(NO₃⁻)的持续排放已严重破坏了这一自然平衡,危及饮用水安全,并对人类健康构成直接威胁—这是亟待解决的跨界环境挑战。尽管传统的化学和生物反硝化技术可以部分缓解污染,但其普遍受限于高成本、低能效以及二次污染的潜在危险。在此背景下,电催化硝酸盐还原(NO₃RR)应运而生,成为一种有前景的可持续氮管理策略,其利用可再生电力将废水中的NO₃⁻污染物转化为有价值的氨(NH₃)。相较于Haber-Bosch工艺理论上最低所需的13 kWh kg⁻¹能耗,NO₃RR提供了卓越能效和环境相容性。至关重要的是,在中性pH条件下运行NO₃RR能高度模拟现实世界的水生环境,从而实现同步NO₃⁻修复和选择性NH₃合成—为可持续氮管理提供了一种实用、环境友好且经济可行的解决方案。
然而,由于N=O键的惰性和缓慢的质子耦合电子转移动力学,NO₃RR面临高过电位的问题,导致过度能耗(>20 kWh kg⁻¹)和低效率。阴极界面过高的质子浓度会促进竞争性析氢反应(HER),而缺质子环境则会限制氢化步骤;同时,外部施加的酸性或碱性条件容易导致催化剂失活,损害反应器的耐久性和可扩展性。催化剂活性位点的协同调节和界面碱性的动态维持,为活化NO₃⁻、平衡质子可用性并抑制HER提供了有效策略。局域碱性微环境不仅能抑制副产H₂析出,还能加速质子耦合电子转移(PCET)动力学。然而,当前策略主要依赖于静态化学修饰—例如,接枝胺或氟官能团、掺杂金属氢氧化物以创建局域碱性区域,缺乏动态适应性。在中性电解液中,界面水刚性的氢键网络进一步阻碍了质子传输和水解离,而后者是质子的主要来源。因此,在实际条件下,动态产生并维持界面碱性的能力仍然是提升NO₃RR动力学的核心挑战。
在此,该研究介绍了一种由MnFe双位点氧化物实现的“自维持碱性局域微环境”策略,该氧化物同时作为结构支架和催化介质,其中FeOₓ中无活性FeOh位点被Mn选择性取代,而活性FeTd位点得以保留。该设计克服了中性pH下NO₃RR中动力学迟缓和严重HER的持续挑战,消除了对强碱性电解液的传统依赖。一维MnFeOₓ中的Fe位点活化NO₃⁻,并在施加电位下动态将吸附的OH⁻转化为FeOOH,在电极-电解液界面建立起一个动态碱性界面区域,有效抑制了HER。同时,Mn位点稳定高价态Fe物种,并持续将界面H₂O解离为OH⁻和H*,确保了该碱性微环境的稳健维持。所得一维MnFeOₓ催化剂在中性介质中实现了95.9%的NH₃法拉第效率(12.3 mg h⁻¹cm⁻²),并能稳定运行超过20小时而无性能衰减。该研究将局域pH调控从外部干预推进到智能自调节,为超越NO₃RR的自适应电催化剂设计以及在实用水环境中实现高效污染物去除和增值化学品合成提供了新见解。
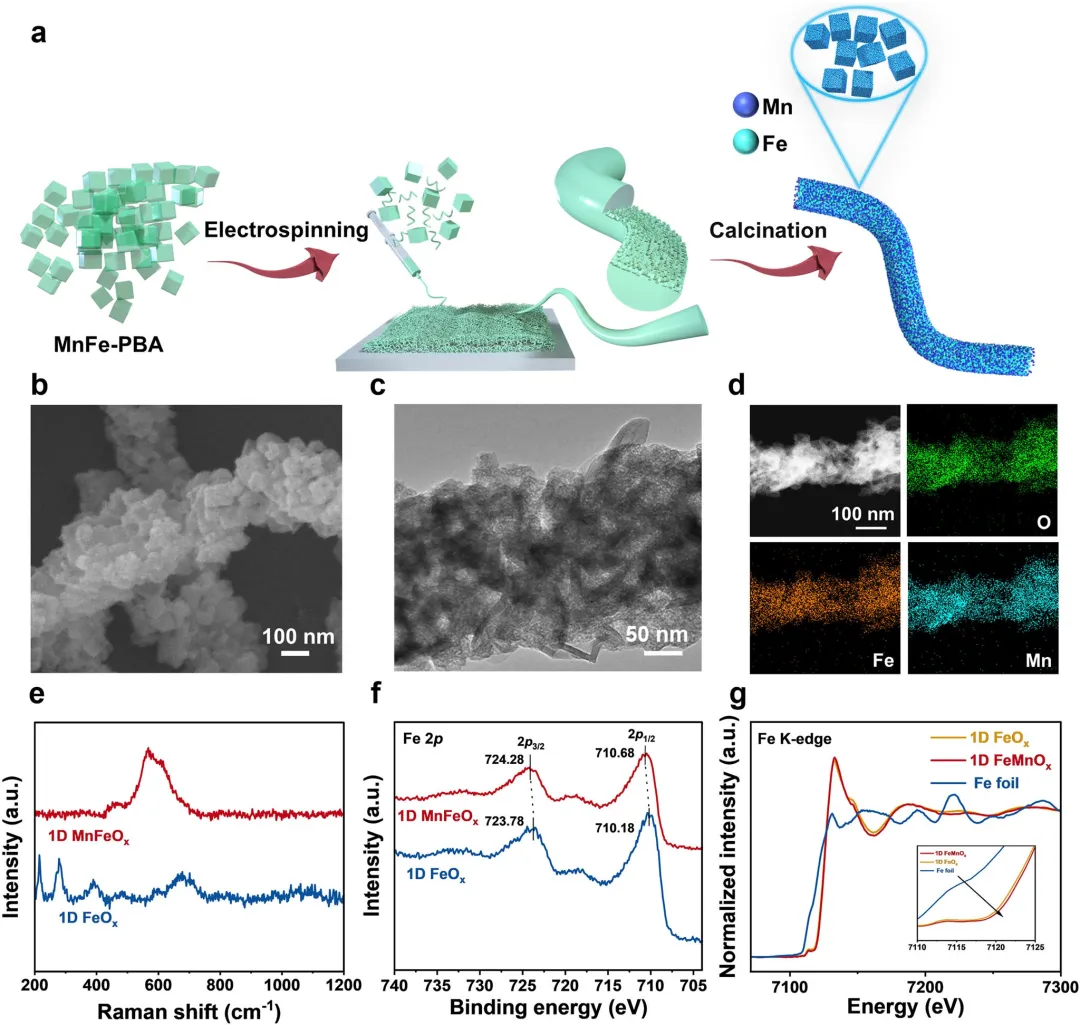
图1 | (a) 一维MnFeO合成过程示意图。(b) 一维MnFeO的场发射SEM图像。(c,d) 一维MnFeO的HRTEM图像和HRTEM-EDS元素映射图像。(e) 一维MnFeO和一维FeO的拉曼光谱。(f) 一维MnFeO和一维FeO的Fe 2p XPS光谱。(g) 一维MnFeO和一维FeO在Fe K边的归一化XANES光谱,以标准Fe箔作为参考。
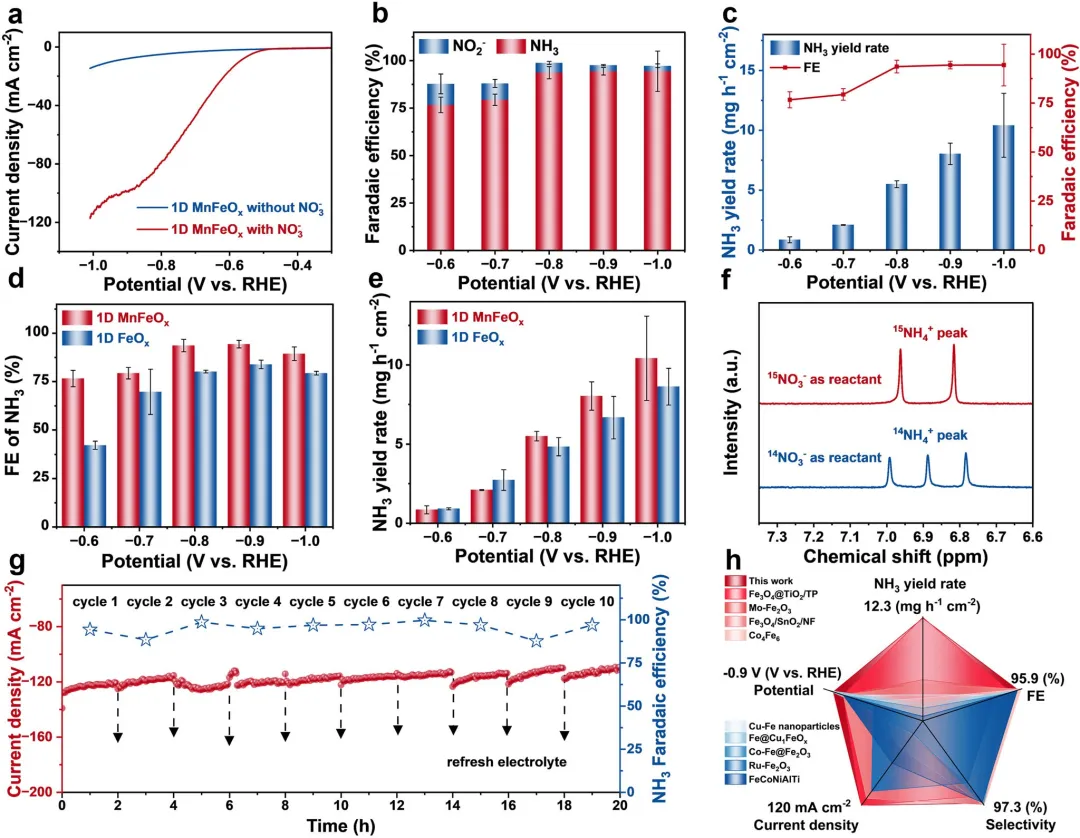
图2 | (a) 一维MnFeO在含有和不含0.05 M KNO₃的0.5 M K₂SO₄电解液中的LSV曲线。(b) 一维MnFeO在给定电位下的FE值。(c) 一维MnFeO的NH₃产率和FE。(d) 不同催化剂在给定电位下的FE比较。(e) 不同催化剂在给定电位下的NH₃产率比较。(f) 使用K¹⁴NO₃和K¹⁵NO₃进行NO₃RR后电解液的¹H NMR谱。(g) 在含有0.05 M KNO₃的0.5 M K₂SO₄电解液中,于-0.9 V vs. RHE电位下进行十次连续循环测试电化学NO₃RR制NH₃的电流密度和FE评估。(h) 本工作中一维MnFeOₓ的FE(%)、NH₃选择性(%)、NH₃产率(mg h⁻¹cm⁻²)、电流密度(mA cm⁻²)和电位(V vs. RHE)与近期报道的中性介质中用于NO₃RR活性的铁基电催化剂的比较。
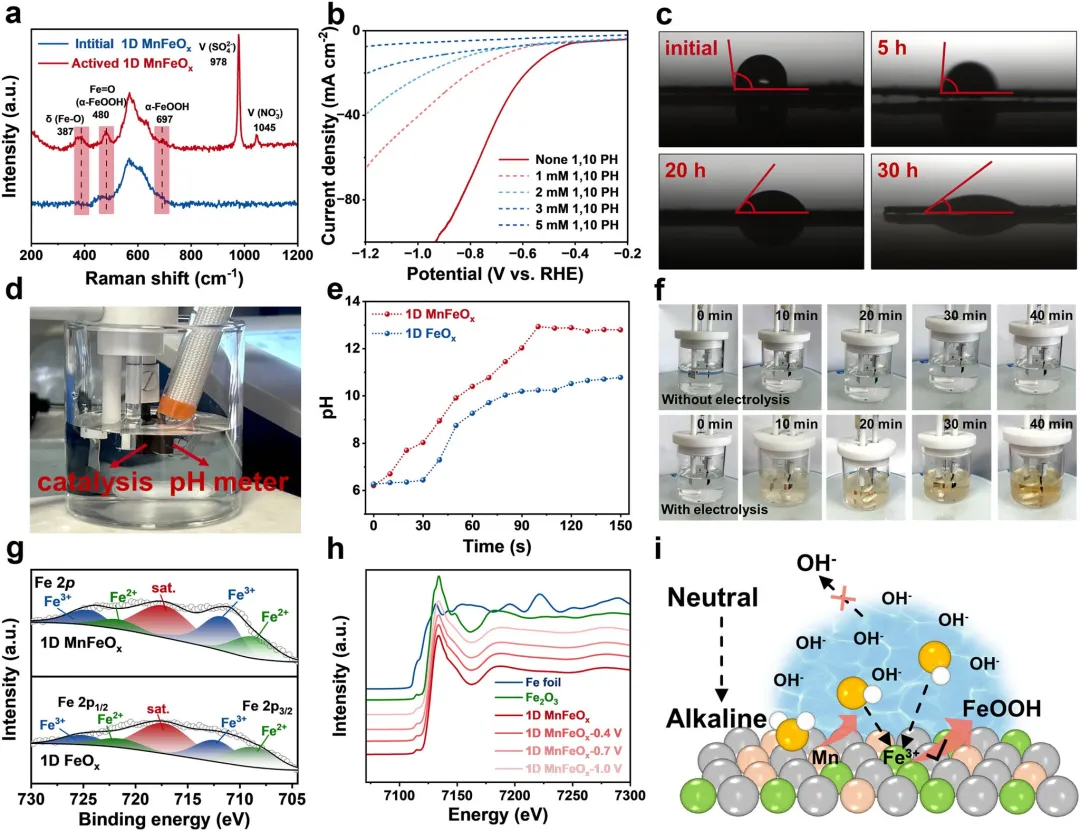
图3 | (a) 初始和活化后的一维MnFeOₓ的拉曼光谱。(b) 一维MnFeOₓ在含有0.5 M K₂SO₄和0.05 M KNO₃及不同浓度1,10-菲咯啉(1,10-PH)作为清除剂的溶液中的LSV曲线。(c) 不同电解时间下一维MnFeOₓ的接触角测量。(d) 使用pH计进行原位pH测量的照片。(e) 使用pH计方法测得的一维MnFeOₓ和一维FeOₓ表面pH值随反应时间的变化。(f) 0.5 M K₂SO₄电解液(含0.05 M KNO₃和3 mM 1,10-PH)的颜色演变。上:电解条件下;下:非电解条件下。(g) 在-0.9 V vs. RHE下电解5小时后一维MnFeOₓ和一维FeOₓ催化剂的高分辨率Fe 2p XPS谱。(h) 一维MnFeOₓ在不同电位下的原位XANES谱。(i) 一维MnFeOₓ阴极表面自维持局域碱性生成示意图。
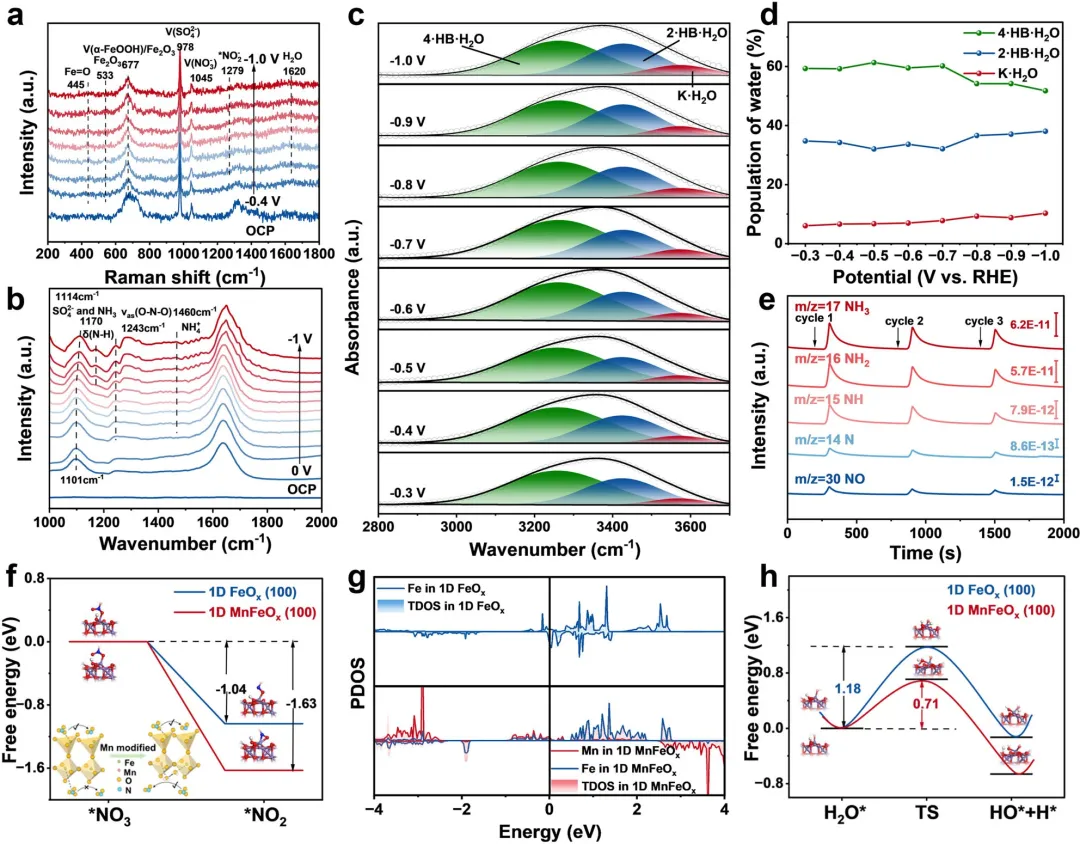
图4 | (a) 一维MnFeO在不同电位下的原位拉曼光谱。(b) 一维MnFeO在不同电位下的原位ATR-FTIR光谱。(c) 一维MnFeO表面界面水O-H伸缩模式的原位ATR-FTIR光谱。(d) 通过原位ATR-FTIR光谱测得的一维MnFeO表面界面水的电位依赖分布。(e) 一维MnFeO上电催化NO₃RR的原位DEMS测量。(f) 一维MnFeO和一维FeO上NO₃RR的决速步吸附能。(g) 一维MnFeO和一维FeO中Fe和Mn轨道的PDOS图。(h) 一维MnFeO和一维FeO上H₂O的吸附能。
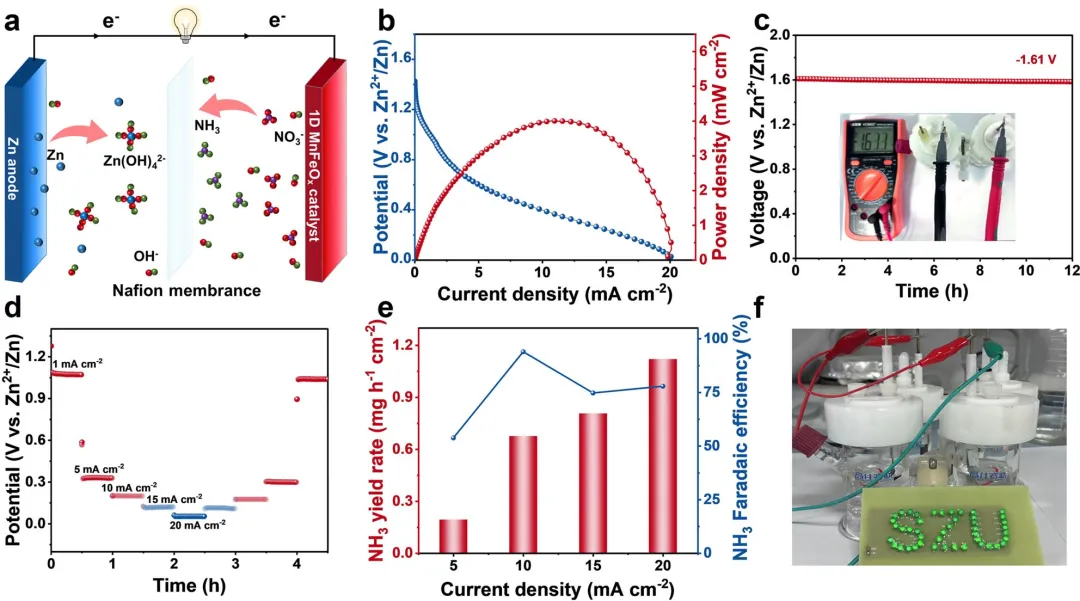
图5 | Zn-NO₃⁻电池性能。(a) 示意图。(b) 放电性能和相应的功率密度。(c) 基于一维MnFeOₓ的Zn-NO₃⁻电池的开路电压。(d) 基于一维MnFeOₓ的Zn-NO₃⁻电池在各种指定电流密度下的恒流放电曲线。(e) 一维MnFeOₓ在不同电流密度下的NH₃产率和FE。(f) 由两个基于一维MnFeOₓ的Zn-NO₃⁻电池供电的灯泡照片。
总之,该研究引入了一种创新的自维持碱性微环境机制,以克服中性pH下NO₃RR中存在的动力学迟缓和竞争性HER的限制,并消除了对高pH电解液的传统依赖。一维MnFeOₓ双位点催化剂同时作为结构支架和功能介质,在电极-电解液界面建立并维持局域高pH微环境。原位拉曼光谱证实了FeOOH物种的稳定生成,而pH测量显示一维MnFeOₓ表面的pH值高达12.93。选择性中毒实验进一步证明,由FeOOH创造的碱性环境通过调节界面质子转移促进了NO₃RR。密度泛函理论DFT计算表明,Mn位点不仅能稳定高价态Fe物种并促进FeOOH形成,还能作为活性中心持续解离界面水(H₂O→OH⁻+ H*),补充OH⁻以维持FeOOH循环。该集成策略为调控自维持碱性微环境以加速中性条件下NO₃RR和绿色NH₃合成提供了一条新途径。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
