丨旨在分享学术交流丨能力有限欢迎指正丨
「上海岱算科技有限公司」已向境内外230余家高等院校/科研院所提供了累计1400多项模拟计算服务,赋能科学研究提速增效!合作实验课题组在线发表学术论文期刊有ACS系列、AM系列、Angew、CEJ、EST、JACS、Matter、Nature子刊等,助力科研工作锦上添花!电化学CO₂还原(ECR)制甲酸盐为可持续化学品生产提供了一条途径,但其工业化性能受到高电流密度下质子供应有限的制约。以往的研究主要关注催化剂的电子结构与CO₂活化过程,而界面水构型在调控水解离动力学中的作用却被忽视。
2026年03月18日,深圳大学骆静利、刘绍庆、吴淑雯团队在Advanced Functional Materials期刊发表题为“Engineering Hydrogen-Down Water Configurations for Ampere-Level Electrochemical CO2 Reduction to Formate”的研究论文,团队成员Guo Xiao-Dong为论文第一作者,骆静利、刘绍庆、吴淑雯为论文共同通讯作者。
第一作者:Guo Xiao-Dong
通讯作者:骆静利、刘绍庆、吴淑雯
通讯单位:深圳大学
论文DOI:10.1002/adfm.74953
该研究利用HfO₂作为Bi表面分子开关,调控界面水分子取向,使其倾向于氢朝下(OH₂↓)构型而非氧朝下(H₂O↓)构型。通过从头算分子动力学AIMD模拟和密度泛函理论DFT计算证实,OH₂↓构型显著降低了水解离能垒,从而产生活性氢物种。多种原位表征手段验证了界面水结构的变化。HfO₂修饰的Bi催化剂实现了优异的甲酸盐部分电流密度(–970 mA cm⁻²)和97%的法拉第效率。在膜电极组件中,该催化剂在0.2–1.6 A的电流范围内表现出高甲酸盐选择性(>90%),确立了界面水工程作为一种有前景的工业化ECR催化剂设计策略。
对可持续能源解决方案的迫切需求推动了电化学CO₂还原(ECR)生产高值化学品的广泛研究。在众多ECR产物中,甲酸因其显著的经济优势以及在化学制造、储能、氢载体和制药工业中的广泛应用而脱颖而出。然而,实现工业级ECR性能仍具挑战性,尤其是在高电流密度条件下,质子供应成为限制因素。虽然传统的ECR研究聚焦于催化剂的电子结构和CO₂活化机理,但近年来,研究重心已转向水分解和质子化过程,因其在多步质子耦合电子转移反应中起着关键作用。
水在电催化反应中的参与主要通过三种途径进行研究:促进H₂O分解过程、调控表面氢键网络以及调控界面水分子构型。前两种途径属于宏观调控策略,并通过本体电解质工程和表面修饰在ECR领域取得了显著进展。然而,对水分子取向的微观调控——这一决定水解离动力学的基本因素——在ECR体系中仍未得到充分探索,尽管在其他电催化反应中已有广泛研究。特定的界面水构型,包括氢朝下水(OH₂↓)、氧朝下水(H₂O↓)和平行水(H₂O=)对ECR性能的影响尚不清楚。
近期关于析氢及其他电催化体系的研究表明,与H₂O↓取向相比,OH₂↓构型表现出显著更低的解离能垒,这有利于快速质子生成并提升催化活性。这种分子层面的理解表明,调控界面水取向可能是增强ECR性能的一种有效策略。具有强亲氧性的过渡金属氧化物在电化学条件下易于原位形成吸附态羟基物种。这些表面键合的羟基基团能够通过氢键相互作用调节局部水分子的取向,可能引导界面水朝向有利于解离和质子供给的OH₂↓构型转变。在此,该研究专注于调控界面水形成微观OH₂↓构型,从而促进ECR加氢反应,实现甲酸盐的工业化规模生产。选择HfO₂作为控制界面水构型的分子开关,因其具有较强OH结合亲和力。分子动力学MD模拟显示,通过HfO₂修饰Bi表面,由于Hf–OH物种与水分子之间的氢键相互作用,驱动水分子构型从H₂O↓取向显著转变为OH₂↓取向。OH₂↓构型显著降低了水解离的能垒,生成活性氢物种,从而有望增强加氢活性。
为验证这一概念,研究人员通过两步法合成了负载在多孔碳上的HfO₂修饰Bi催化剂。多种表征技术证实,HfO₂修饰确实改变了ECR过程中的界面水构型,增加了自由水分子数量,同时减少了氢键结合水。计算与实验分析均表明,与未修饰的催化剂相比,在修饰后的表面上,水解离产生活性氢的反应更有利,且CO₂加氢能垒降低。通过利用这一稳定的质子源,该催化剂实现了优异的甲酸盐部分电流密度(–970 mA cm⁻²),同时保持了97%的法拉第效率。在膜电极组件中,该催化剂在宽电流范围(0.2–1.6 A)内对ECR制甲酸盐表现出高选择性(>90%),并且稳定运行超过60小时。该研究证明,调控微观水取向是开发工业级ECR催化剂的一条有前景的途径。
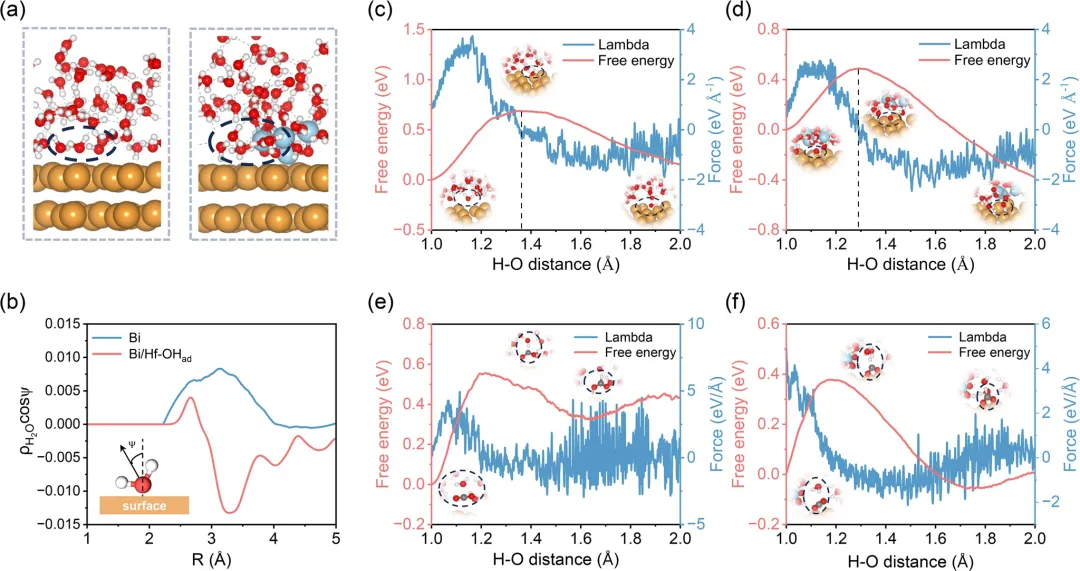
图1 采用动态取向水分子法,模拟得到的Bi表面和Bi/Hf–OHad表面界面结构快照。(b) Bi和Bi/Hf–OHad上方界面水分子偶极取向(ρH₂Ocosψ)随距离的变化,计算值并在10 ps内取平均。(c) Bi和(d) Bi/Hf–OHad上H₂O解离过程的自由能变化与CV的函数关系。(e) Bi和(f) Bi/Hf–OHad上CO₂加氢过程的自由能变化与CV的函数关系。
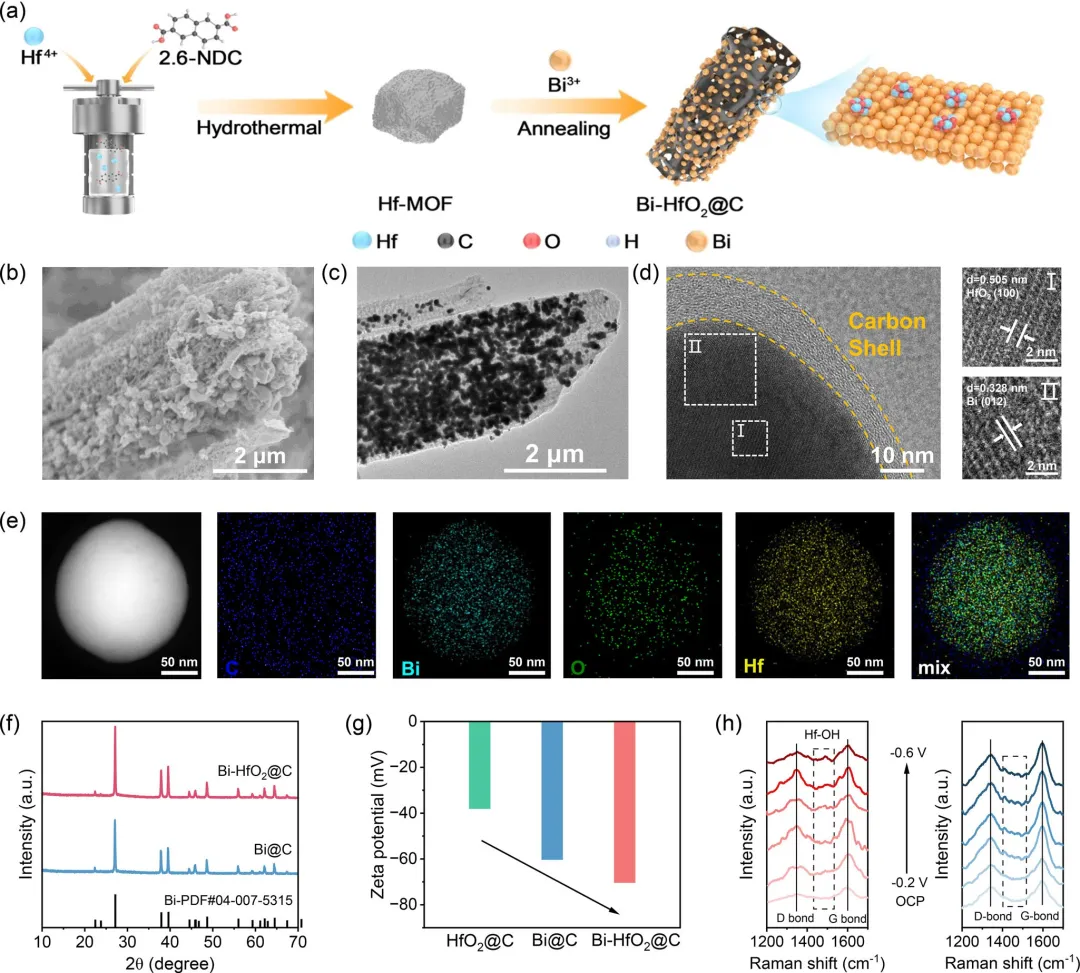
图2 (a) Bi-HfO₂@C合成过程示意图。(b) Bi-HfO₂@C的SEM和 (c) TEM图像。(d) Bi-HfO₂@C的HRTEM图像和 (e) 相应的EDX元素分布图。(f) Bi-HfO₂@C和Bi@C的XRD图谱。(g) Bi-HfO₂@C、Bi@C和HfO₂@C的Zeta电位值。(h) Bi-HfO₂@C和Bi@C在ECR过程中不同电位下的原位拉曼光谱。
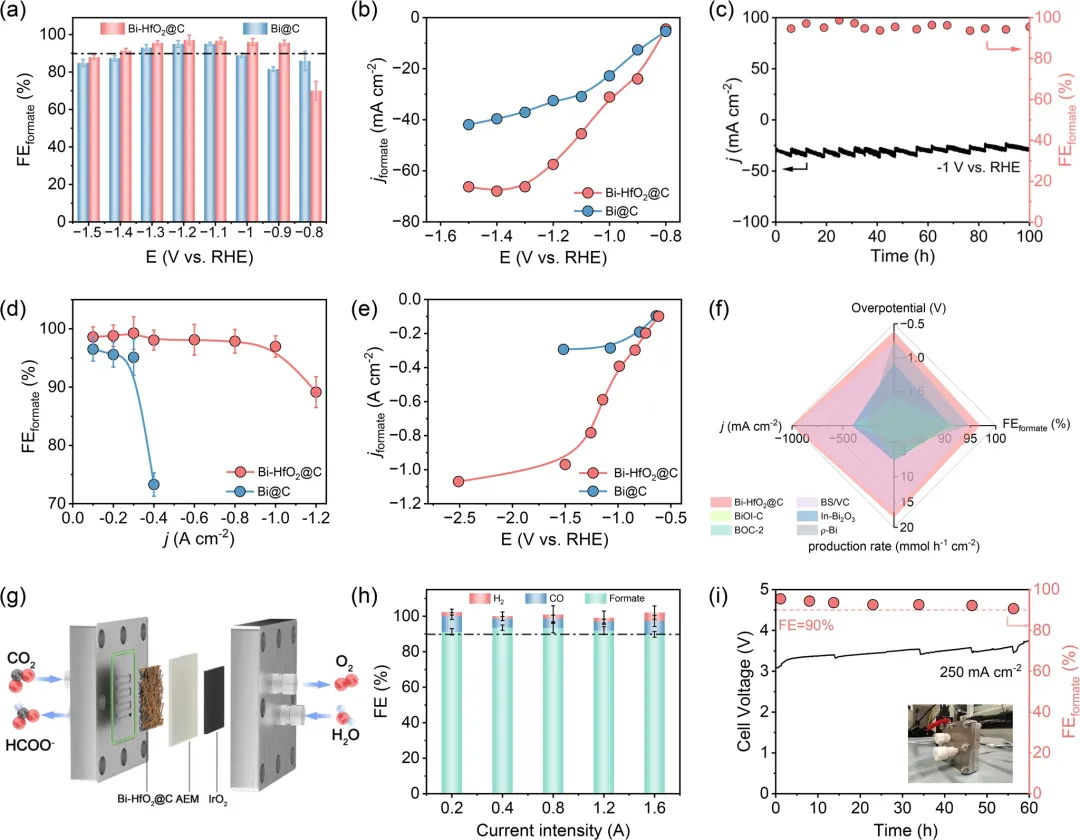
图3 (a) Bi-HfO₂@C和Bi@C在H型电解池中、CO₂饱和的0.5 M KHCO₃溶液中的甲酸盐法拉第效率和 (b) 甲酸盐分电流密度。(c) Bi-HfO₂@C在H型电解池中–1 V下的稳定性测试。(d) Bi-HfO₂@C和Bi@C在流动池中、1 M KOH溶液中的甲酸盐法拉第效率和 (e) 甲酸盐分电流密度。(f) Bi-HfO₂@C与近期报道的Bi基电催化剂在200 mA cm⁻²电流密度下的过电位、甲酸盐法拉第效率及产率性能比较。(g) 采用阴离子交换膜电解槽(2×2 cm²)的膜电极组件构型示意图。(h) 在膜电极组件电解槽中、1 M KOH溶液下,不同施加电流强度下的法拉第效率。(i) Bi-HfO₂@C在膜电极组件电解槽中的长期稳定性测试。
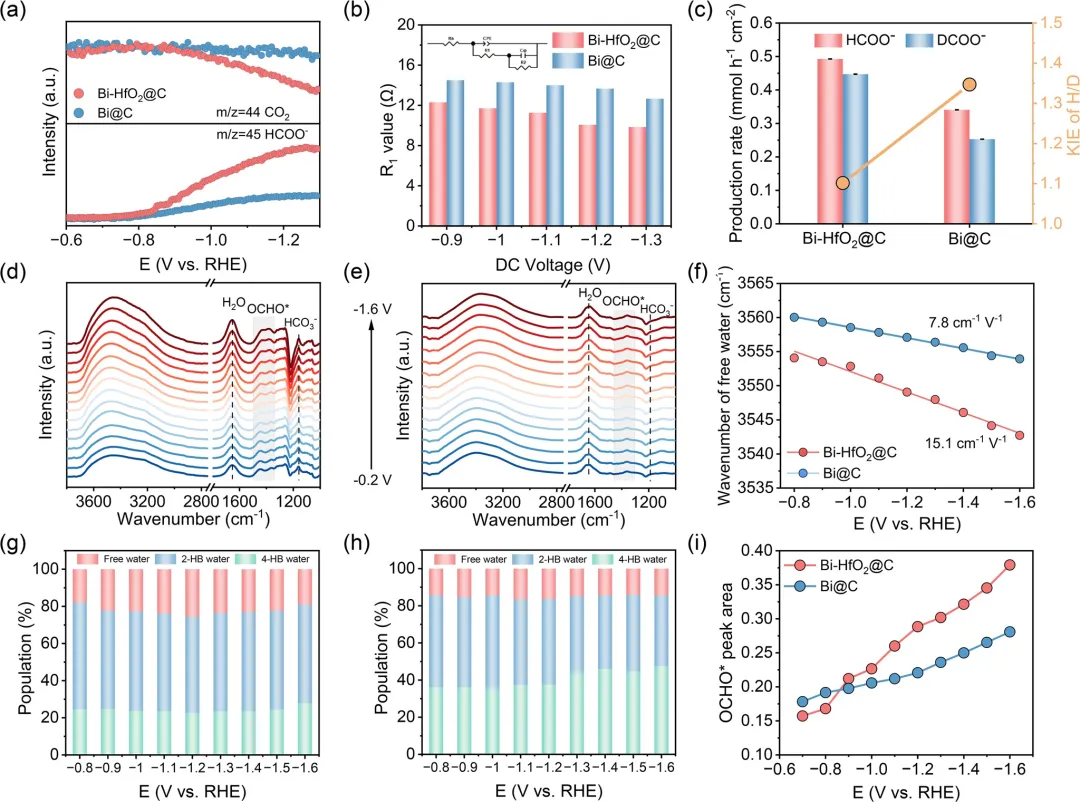
图4 (a) Bi-HfO₂@C和Bi@C在ECR制甲酸盐过程中的在线DEMS分析。(b) 从Bi-HfO₂@C和Bi@C在1 M Ar饱和KOH溶液中测试的奈奎斯特图推导出的不同直流电压下的R₁值比较。(c) Bi-HfO₂@C和Bi@C在–1.1 V vs. RHE下由甲酸盐生成速率推导出的H/D动力学同位素效应。在CO₂饱和的0.5 M KHCO₃溶液中记录的 (d) Bi-HfO₂@C和 (e) Bi@C的原位ATR-SEIRAS光谱。(f) Bi-HfO₂@C和Bi@C上自由水的O-H伸缩振动斯塔克调谐率。(g) Bi-HfO₂@C和 (h) Bi@C在不同电位下界面水比例的变化。(i) Bi-HfO₂@C和Bi@C在不同电位下OCHO中间体的峰面积。
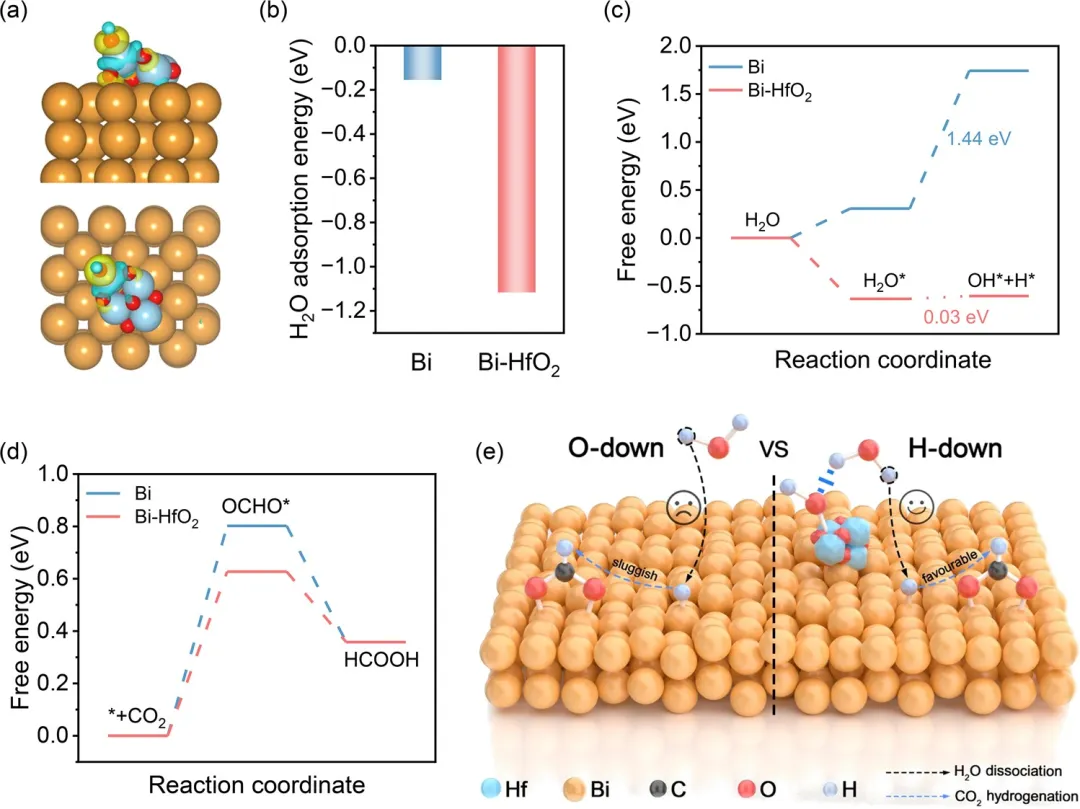
图5 (a) Bi-HfO₂在不同视角下吸附OH的电荷密度差计算。(b) Bi和Bi-HfO₂上的H₂O吸附能。(c) Bi-HfO₂和Bi上H₂O解离过程的吉布斯自由能图。(d) Bi-HfO₂和Bi上HCOOH生成的吉布斯自由能图。(e) Bi-HfO₂上OH₂↓解离以及Bi上H₂O↓解离的示意图。
总之,该研究报道了通过微观水取向工程实现工业化规模的ECR制甲酸盐电解。AIMD模拟、DFT计算和原位SEIRAS测量表明,在Bi表面修饰HfO₂可将界面水从氧朝下(H₂O↓)取向调控为有利的氢朝下(OH₂↓)取向。这导致Volmer步骤的活化能垒显著降低,确保了在高电位下高效、持续的氢供应以驱动CO₂加氢。因此,Bi-HfO₂催化剂在1 A cm⁻²电流密度下实现了高的甲酸盐法拉第效率(97%),性能优于目前报道的大多数Bi基催化剂。在膜电极组件中,该催化剂在宽电流范围(0.2至1.6 A)内对ECR制甲酸盐表现出高选择性(>90%),并且稳定运行超过60小时。该研究揭示了界面水取向在实现工业化规模CO₂电解中的关键作用,并有望推广应用于其他涉及加氢过程的催化反应。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
