「上海岱算科技有限公司」已向境内外230余家高等院校/科研院所提供了累计1400多项模拟计算服务,赋能科学研究提速增效!合作实验课题组在线发表学术论文期刊有ACS系列、AM系列、Angew、CEJ、EST、JACS、Matter、Nature子刊等,助力科研工作锦上添花!在水系电解液中进行醛的电化学氨氧化反应,为腈类化合物的合成提供了一条可持续且极具前景的途径。然而,该反应的效率,特别是对腈的选择性,受到醛直接氧化竞争反应的限制,该竞争反应由醛自身不可避免且不可控的水合作用引发。
2026年04月15日,中国科学技术大学熊宇杰、张宁团队在Journal of the American Chemical Society期刊发表题为“The Salting-Out Effect Enables Highly Selective Electrochemical Ammoxidation of Aldehydes to Nitriles”的研究论文,中科大Liu Zhenzhong/Liu Yijun、松山湖材料实验室Cai Lejuan为论文共同第一作者,熊宇杰、张宁、中国科学院深圳先进技术研究院商健为论文共同通讯作者。
第一作者:Liu Zhenzhong、Liu Yijun、Cai Lejuan
通讯作者:熊宇杰、张宁、商健
通讯单位:中国科学技术大学、中国科学院深圳先进技术研究院
论文DOI:10.1021/jacs.6c01267
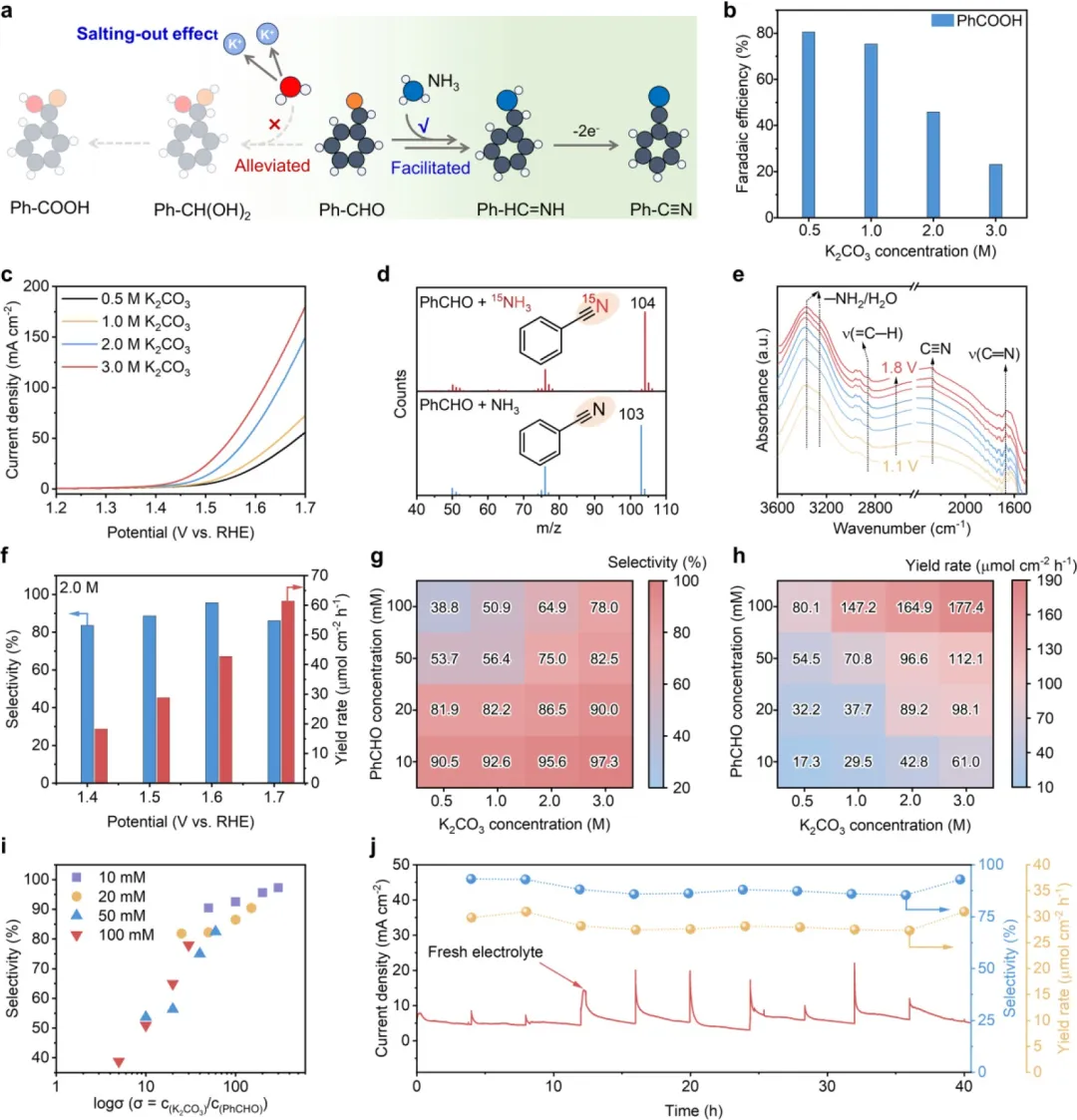
该研究展示了由体相电解液中高浓度钾离子(K⁺)诱导产生的盐析效应,能够实现高效腈类化合物合成。分子动力学MD模拟和光谱学研究表明,高浓度K⁺促使水(H₂O)结构重取向,从而主导更强的K⁺-H₂O离子-偶极相互作用。这种溶剂微环境的调控反过来减弱了H₂O对醛的亲和力,从而抑制了醛的水合反应。因此,醛与氨的缩合反应得到促进,有利于腈的生成。以苯甲醛为概念验证底物,在浓度为3.0M的K₂CO₃电解液中,苯甲腈的产量显著提升。当分别加入10mM和100mM苯甲醛时,可获得高达97.3%的选择性和177.4μmol cm⁻² h⁻¹的可观产率。这种由盐析效应诱导的选择性提升还表现出良好的反应耐久性和醛类底物普适性,彰显了其在可持续化学品制造方面的巨大优势。
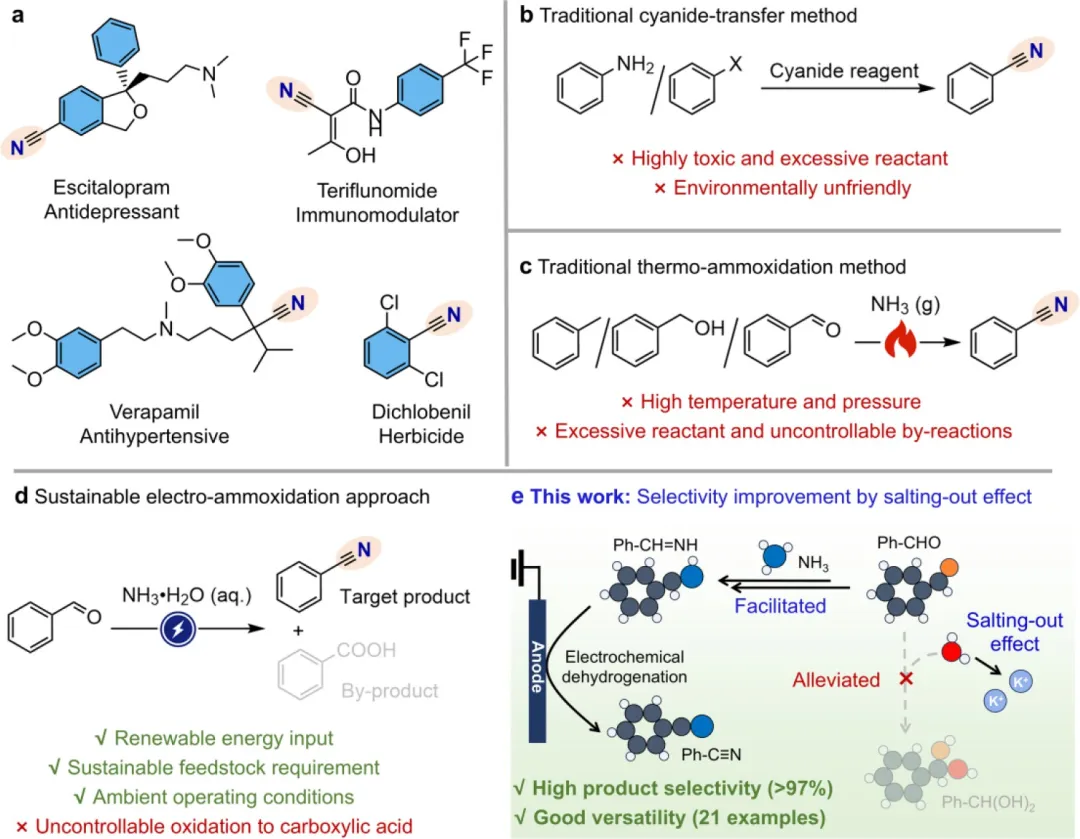
腈类化合物通常表现出良好的生物正交反应活性,可作为多种精细化学品(包括药物和农用化学品)的核心构建模块,如图1a选择性所示。鉴于其关键重要性,研究人员已开发出多种合成方法,以便根据可获得的原料来获取所需的腈类化合物。氰基转移反应,例如Sandmeyer反应或Rosenlund-von Braun反应,是一类有效的工艺,但必需且过量使用有毒氰化物盐通常会引发对环境完整性的严重担忧(图1b)。为了解决这一问题,低毒的氨源(NH₃)通过烃、醇和醛等碳原料的氨氧化反应合成腈类化合物而受到特别关注(图1c)。然而,该路线存在条件苛刻(高温高压)以及原子经济性低(过量氧化剂和不可控副产物)的问题。在这方面,开发高效且可持续的创新合成方法尤为理想。
近年来,由于内在优点(包括温和条件和高可持续性),利用电合成方法生产精细化学品取得了显著进展。使用醇/醛和氨水溶液(aq. NH₃)作为原料,通过选择性C-N偶联反应,已成功合成了多种有机氮化合物,包括酰胺和腈。然而,就通过醛氨氧化合成腈(图1d)而言,反应效率仍不理想。从根本上讲,该反应始于醛与氨缩合形成半缩醛胺中间体。因此,这种困境的内在根源在于醛不可避免但不可控的水合反应,生成偕二醇物种,后者在电氧化条件下竞争性地演变为羧酸副产物。为了提高腈的选择性,迫切需要一种能够促进醛与NH₃而非H₂O相互作用的策略。
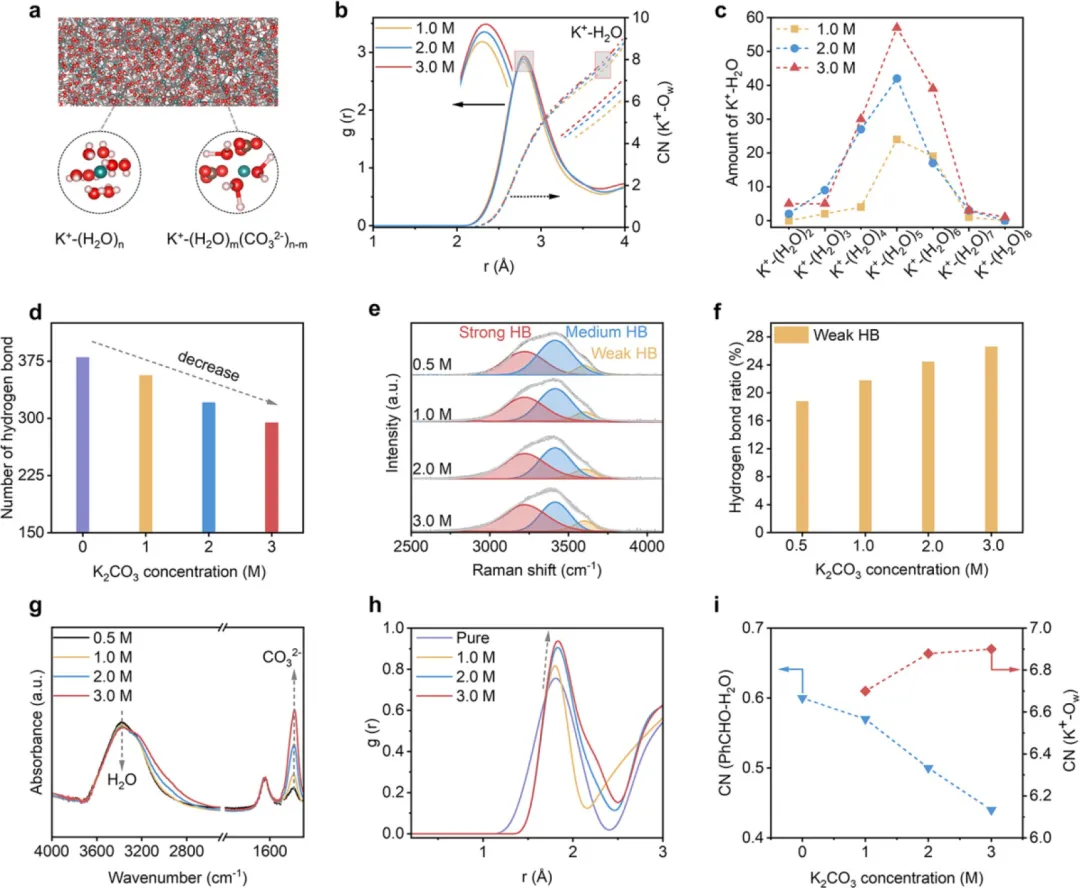
在水系电解液中,H₂O本身在调控溶剂微环境中起着关键作用,它既可以通过溶剂化效应与反应物相互作用,也可以构建多个氢键(HBs)。在这方面,通过调控H₂O的配位相互作用来调节H₂O的活性是特别设想的。一种广泛适用的方法是利用碱金属阳离子(尤其是离子半径大、电荷密度低的阳离子,如K⁺和Cs⁺)诱导的盐析效应。这些阳离子能通过离子-偶极相互作用吸引极性H₂O分子,形成水合层以限制H₂O的重新定向,从而展现出对HB网络的结构破坏特征。这种情况被认为能有效促使H₂O对溶液中其他物种的亲和力降低。在这种情况下,可以设想醛分子反而有更多机会接触NH₃,从而形成所需的腈。
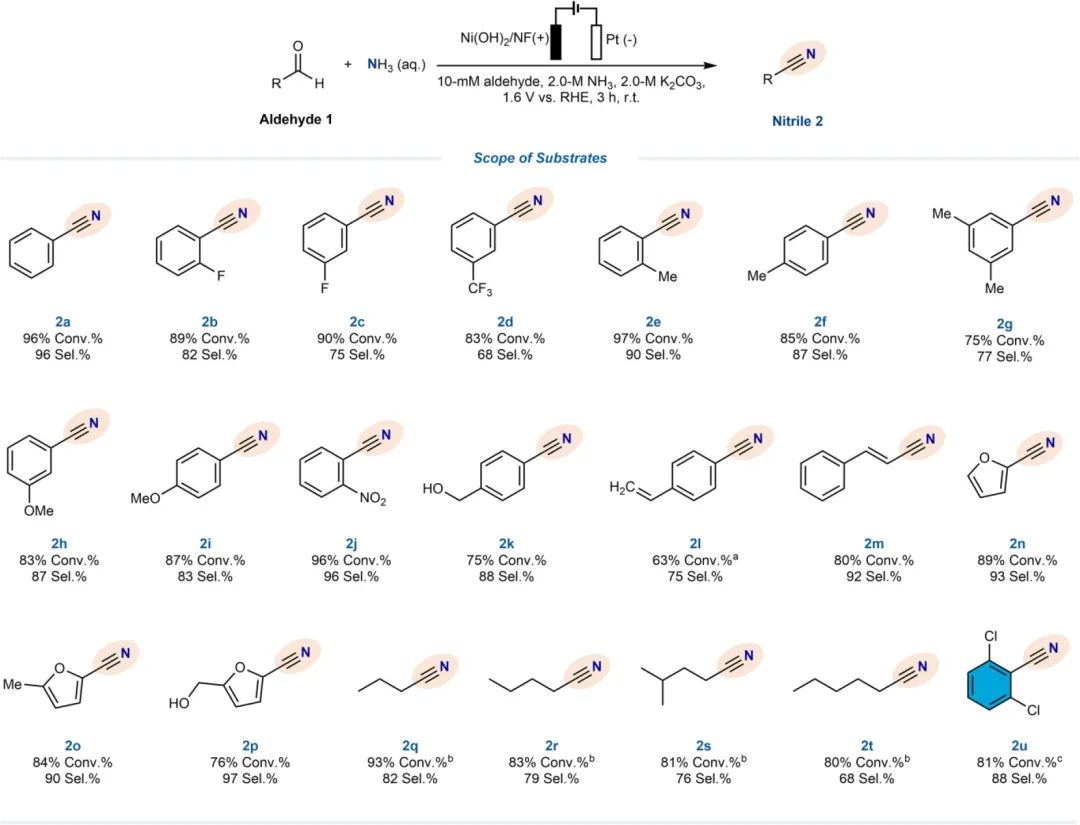
基于这些理解,该研究在此证明了由体相电解液中高浓度钾离子(K⁺)产生的有效盐析效应能够实现醛向腈的高选择性氨氧化反应(图1e)。分子动力学MD模拟和光谱学研究表明,高浓度K⁺促进了阳离子溶剂化,提高了水合层水平以增强K⁺-H₂O相互作用,同时也破坏了HB网络。K⁺诱导的盐析效应降低了水对醛水合反应的活性,因此有效减轻了醛直接氧化为羧酸的过程。同时,醛与NH₃的可及性得到提升,进而通过席夫碱反应促进了亚胺中间体的形成。以苯甲醛(PhCHO)为代表,在含有10mM PhCHO和3.0M K₂CO₃电解液的条件下,施加1.6Vvs RHE的电位,苯甲腈(PhCN)产物的选择性高达97.3%。当PhCHO浓度增至100mM时,该性能依然保持,选择性为78.0%,同时产率可观,达到177.4μmol cm⁻² h⁻¹。底物拓展实验表明,所开发的这种电合成路线对包括芳香醛和脂肪醛在内的21种醛基底物具有优异的普适性,展现出高转化率(63%-97%)和高选择性(68%-97%)。
图1. 不同腈合成方法的比较。(a) 选作药物和农药的腈类化合物。(b, c) 传统的氰基转移法(b)和热氨氧化法(c)用于腈合成。(d) 可持续电氨氧化法用于腈合成的优势与挑战。(e) 本工作提出的通过盐析效应提高腈选择性的策略。
图2. K⁺介导的溶剂化调控研究。(a) 含有不同K⁺溶剂化结构的K₂CO₃电解质示意图。(b) 不同K₂CO₃浓度下K⁺-H₂O相互作用的RDF和配位数。(c, d) 不同K₂CO₃浓度下K⁺-(H₂O)ₙ相互作用模拟量的变化(c)和平均氢键数(d)。(e, f) 不同K₂CO₃浓度下的去卷积拉曼光谱(e)和电解液中以弱氢键为特征的水分子占比(f)。(g) 不同K₂CO₃浓度下电解液的红外光谱。(h, i) 不同K₂CO₃浓度下PhCHO-H₂O相互作用的RDF(h)和配位数(i)。
图3. PhCN电解活性评估。(a) 盐析效应诱导化学平衡促进PhCN形成示意图。(b) 不同K₂CO₃浓度下直接PhCHO电氧化生成PhCOOH的FE。(c) 存在10mM PhCHO和2.0M NH₃时,不同K₂CO₃浓度下的LSV极化曲线。(d) ¹⁵N同位素标记实验中收集到的PhCN产物的MS谱图。(e) PhCN形成过程中在不同施加电位下的电化学原位ATR-SEIRAS谱图。(f) 不同施加电位下PhCN产物的选择性和产率。反应条件:10mM PhCHO,2.0M NH₃,2.0M K₂CO₃电解液,反应时间3小时。(g, h) 不同PhCHO原料和K₂CO₃电解液浓度下PhCN产物的选择性(g)和产率(h)。反应条件:2.0M NH₃,相对于RHE 1.6V,反应时间3小时。(i) PhCN选择性与浓度比率因子(σ)的内在相关性。(j) 连续40小时操作内的反应耐久性测试。反应条件:10mM PhCHO,2.0M NH₃,2.0M K₂CO₃电解液,1.5V vs RHE 。
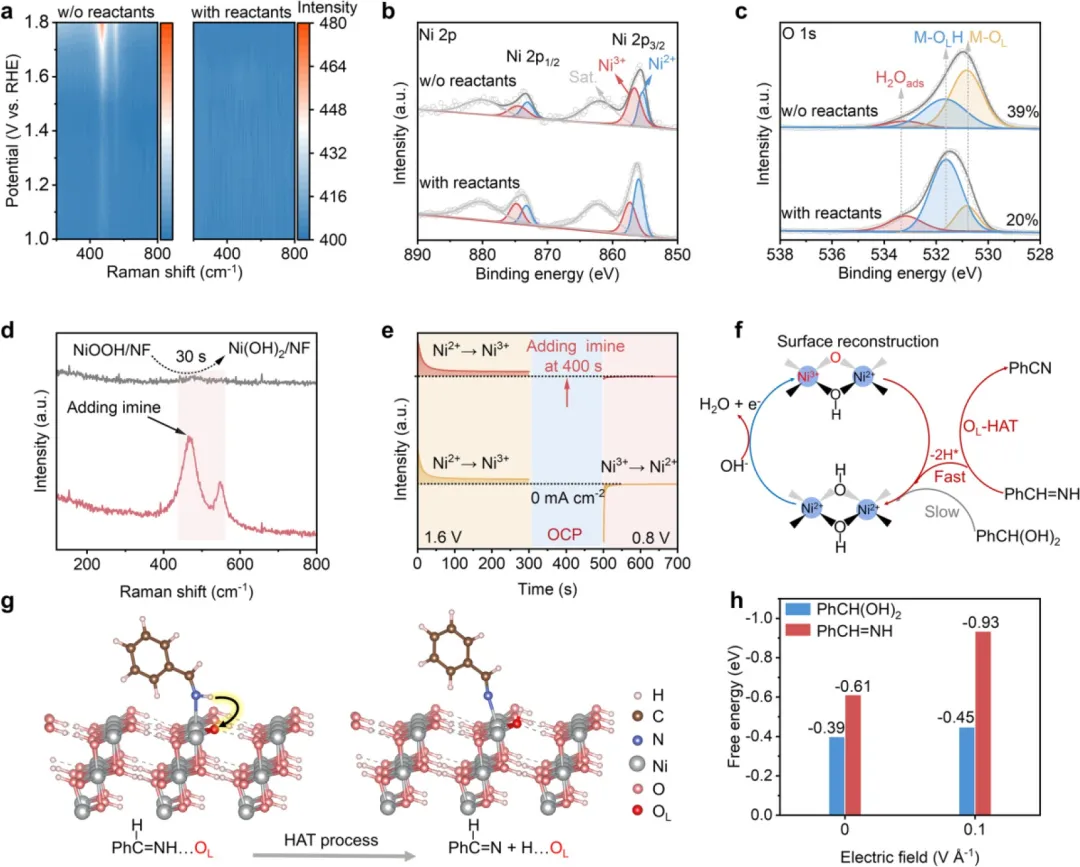
图4. 电化学过程的机理研究。(a) 不存在(左图)和存在(右图)反应物时Ni(OH)₂/NF催化剂的原位拉曼光谱。(b, c) 不存在和存在反应物时,电化学处理后Ni 2p (b) 和O 1s (c) XPS核心能级谱图。(d) 在开路条件下加入亚胺溶液后,生成的NiOOH的拉曼光谱信号演变。(e) Ni(OH)₂电极在1.6V vs. RHE(0-300秒)、开路(300-500秒)和0.8V vs. RHE(500-700秒)顺序条件下的多电位阶跃计时电流曲线。(f) 提出的Oₗ介导的HAT反应路径,用于亚胺脱氢生成腈产物。(g) 亚胺物种向NiOOH层表面亲电Oₗ位点发生HAT过程的模拟示意图。(h) 确定的PhCH=NH和PhCH(OH)₂中间体物种上HAT过程的吉布斯自由能变化。
图5. 腈电解的拓展实验。
总之,该研究展示了一种有效策略,通过醛的氨氧化反应提高概念验证型腈电合成的产物选择性。其关键在于利用体相电解液中高浓度K⁺产生的盐析效应来调控溶剂微环境。结合分子动力学MD模拟与光谱学测量,从根本上理解为:在高K⁺浓度情况下,通过离子-偶极相互作用的K⁺-H₂O配位得到加强,这限制了H₂O的自由度,从而削弱了其对醛的亲和力。因此,醛底物更倾向于与NH₃反应,而不是被H₂O水合,从而有效减轻了其直接氧化为羧酸的过程。因此,当使用10mM PhCHO为原料时,在浓度为3.0M的K₂CO₃电解液中实现了高达97.3%的腈选择性。此外,该选择性对100mM的高PhCHO浓度表现出78.0%的良好耐受性,腈的产率达到177.4μmol cm⁻² h⁻¹。值得注意的是,这种电合成方法还展现出对一系列芳香醛和脂肪醛底物的适用性,具有良好的转化率(63%-97%)和选择性(68%-97%)。该研究为实现腈类化合物的可持续和可规模化生产提供了一种策略,并强调了H₂O配位化学和溶剂微环境在水系电化学合成中实现高效率以实现可持续发展方面的核心作用。


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
