作者
贾传义(贵州师范学院)
叶逸凡(中国科学技术大学)
陈辉煌(深圳大学)
鲍骏(中国科学技术大学)
引言
氢气因其高能量密度和零碳排放特性被视为理想的清洁能源替代品。电化学水分解能够将H₂O直接转化为高纯度H₂,是可持续氢生产的最有前景的方法之一。在各种技术中,碱性水电解是目前最具商业可行性和最广泛采用的方法。作为碱性水分解的关键阴极反应,析氢反应(HER)涉及初始的H₂O解离步骤(Volmer步骤),随后是氢演化过程(Heyrovsky或Tafel步骤)。Pt基单原子催化剂被认为是最有效的HER催化剂。然而,Pt单原子在碱性电解质中缓慢的Volmer步骤动力学导致氢中间体(*H)供应不足,这对实现安培级电流密度构成了根本性障碍。相比之下,(过渡)金属氧化物表现出快速的Volmer动力学,能够向相邻的Pt单原子提供H物种,尽管它们遭受缓慢的Heyrovsky或Tafel步骤动力学。因此,将Pt单原子锚定在(过渡)金属氧化物表面代表了设计高性能碱性HER催化剂的合理策略。对于Pt单原子/过渡金属氧化物体系,Pt单原子的氢吸附关键决定了其碱性HER活性。较弱的氢吸附导致*H供应不足,严重限制了后续的H₂生成动力学。相反,较强的氢吸附促进了*H供应,但也导致较弱的H₂脱附和Pt位点中毒。因此,适度的氢吸附对于实现高碱性HER活性至关重要。氧化物上不同锚定位点的性质,包括电子结构和构型,表现出实质性变化。将Pt单原子锚定在过渡金属氧化物的不同位点上提供了调节其氢吸附行为的有效策略。然而,关于氢吸附与单原子锚定位点之间关系的原子级洞察仍然难以捉摸。
核心发现
本研究通过将Pt单原子选择性地锚定在CoOOH的三种不同位点(氧空位位点PtV/CoOOH、三重空位位点PtT/CoOOH和晶格位点PtL/CoOOH),成功调控了它们的氢吸附行为,实现了安培级碱性析氢反应。电化学测量表明,PtT/CoOOH在10 mA cm⁻²电流密度下实现了仅8 mV的过电位,比PtV/CoOOH和PtL/CoOOH分别低70 mV和8 mV,并展现出超过1000小时的长期稳定性。在阴离子交换膜水电解槽(AEMWE)中,集成PtT/CoOOH仅需1.90 V即可达到1.0 A cm⁻²的工业电流密度,并稳定运行1000小时。原位/工况X射线吸收精细结构(XAFS)、环境压力X射线光电子能谱(AP-XPS)、衰减全反射表面增强红外吸收光谱(ATR-SEIRAS)和理论计算共同证明,PtT/CoOOH表现出适度的H₂O解离动力学和近热中性的氢结合能。这种最优的氢吸附促进了平衡的H吸附-H₂脱附动力学,从而实现了优于具有较弱氢吸附的PtV/CoOOH和较强氢吸附的PtL/CoOOH的碱性HER活性。研究不仅开发了用于将单原子锚定到不同位点的精确合成策略,还阐明了位点依赖的氢吸附对催化性能的影响机制。
图文解读
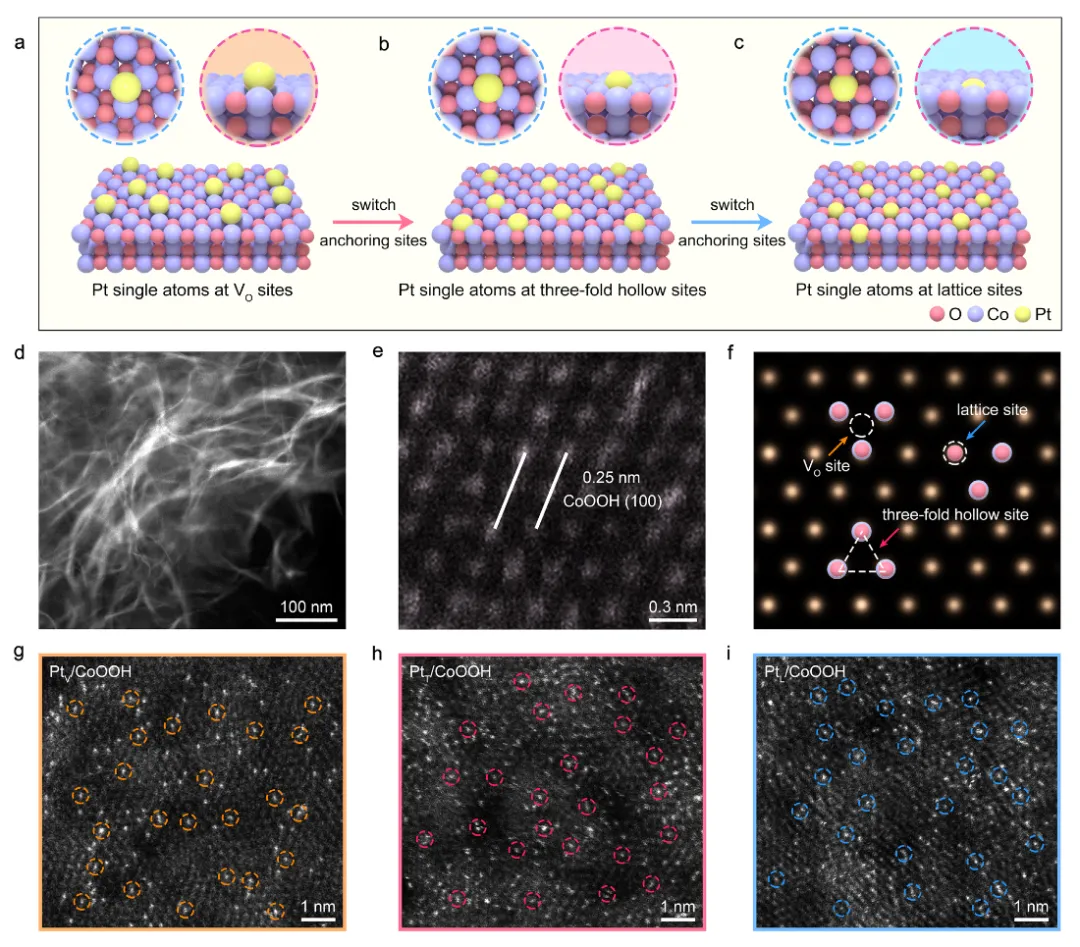
图1:位点特异性Pt单原子的合成和空间分布
图1展示了位点特异性Pt单原子的合成策略和空间分布表征。图1a-c通过示意图展示了Pt单原子在CoOOH三种不同位点的选择性锚定机制:氧空位位点(VO)带正电荷,能够选择性吸附带负电荷的Pt(OH)₆²⁻阴离子;三重空位位点带负电荷,能够选择性吸附带正电荷的Pt⁴⁺离子;晶格位点则通过共沉积方法将Pt单原子引入CoOOH晶格中。图1d-f展示了CoOOH载体的形貌和结构表征,HAADF-STEM图像显示CoOOH为纳米片结构,原子分辨率图像和模拟图像证实了CoOOH表面存在VO位点、三重空位位点和晶格位点。图1g-i展示了三种催化剂的原子分辨率HAADF-STEM图像,通过Z衬度对比可以清晰观察到Pt单原子(亮点)在CoOOH表面的分散情况。通过对比模拟和实验HAADF-STEM图像,精确确定了Pt单原子的锚定位点:PtV/CoOOH中Pt原子位于Co柱的间隙位置,PtT/CoOOH中Pt原子位于三个Co原子形成的三角形间隙内,PtL/CoOOH中Pt原子替代了Co原子进入晶格位置。这些结果为位点特异性单原子的成功合成提供了直接的视觉证据。
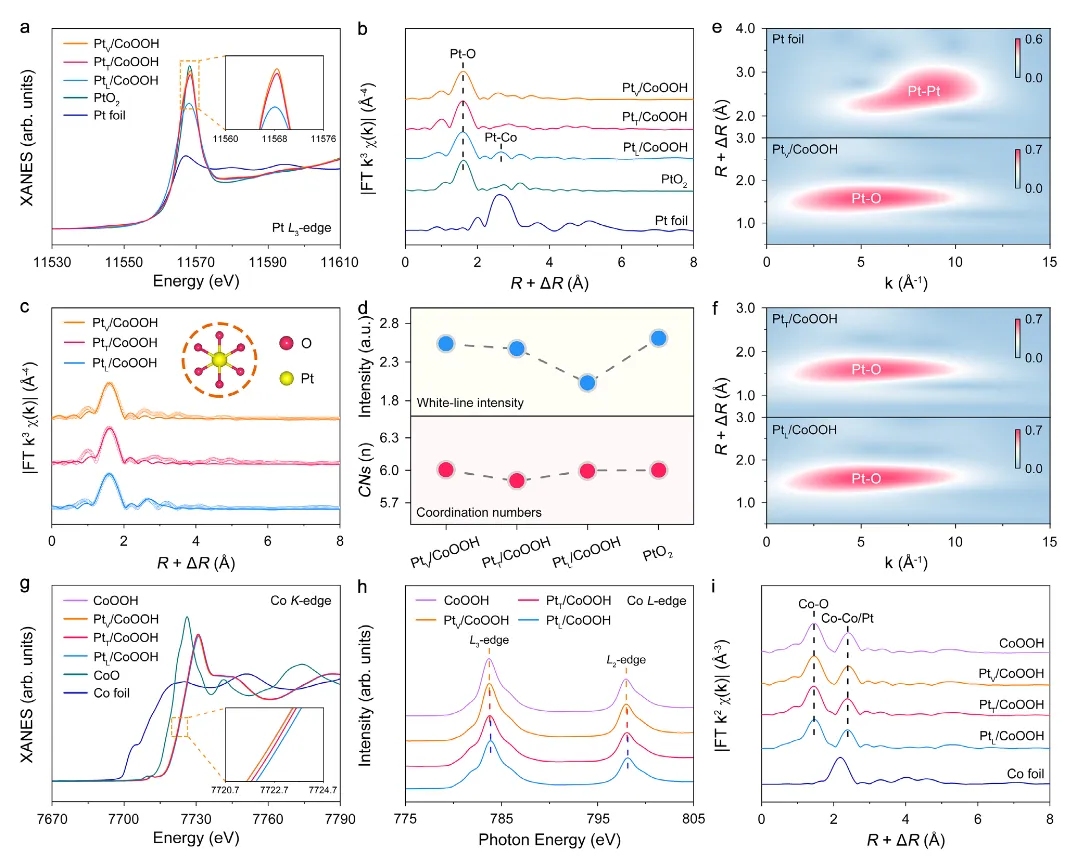
图2:位点特异性Pt单原子的原子结构分析
图2通过X射线吸收谱(XAFS)深入分析了位点特异性Pt单原子的电子结构和配位环境。图2a-d展示了Pt L₃边XANES和EXAFS结果,所有催化剂的白线强度介于Pt箔和PtO₂之间,表明Pt单原子的价态在0到+4之间。白线强度从PtV/CoOOH、PtT/CoOOH到PtL/CoOOH依次降低,表明Pt单原子的价态逐渐降低。EXAFS谱图中,所有催化剂在约1.6 Å处显示出主要峰,归属于第一壳层的Pt-O键合,PtL/CoOOH还在约2.6 Å处显示出归属于Pt-Co键合的小峰,证实Pt单原子被引入CoOOH晶格。EXAFS拟合结果显示所有样品的Pt-O配位数约为6.0,表明Pt单原子形成PtO₆八面体结构。图2e-f的小波变换分析进一步确认了Pt物种的原子分散,所有催化剂在约5.5 Å⁻¹处显示最大强度,对应Pt-O散射,而在约8.6 Å⁻¹处未观察到Pt-Pt散射,证实了Pt物种的孤立分散。图2g-i分析了Co物种的电子结构和配位环境,Co K边XANES显示所有样品的吸收边相对于CoO向高能方向移动,表明Co物种价态高于+2。Co L边软XAS谱图进一步证实了这一结果。EXAFS拟合结果显示CoOOH和PtV/CoOOH的Co-O和Co-Co/Pt配位数分别约为6.0和4.9,而PtT/CoOOH和PtL/CoOOH表现出较低的Co-O配位数(约5.6和5.7)和Co-Co/Pt配位数(约4.7),揭示了位点依赖的Co物种与Pt单原子之间的相互作用。
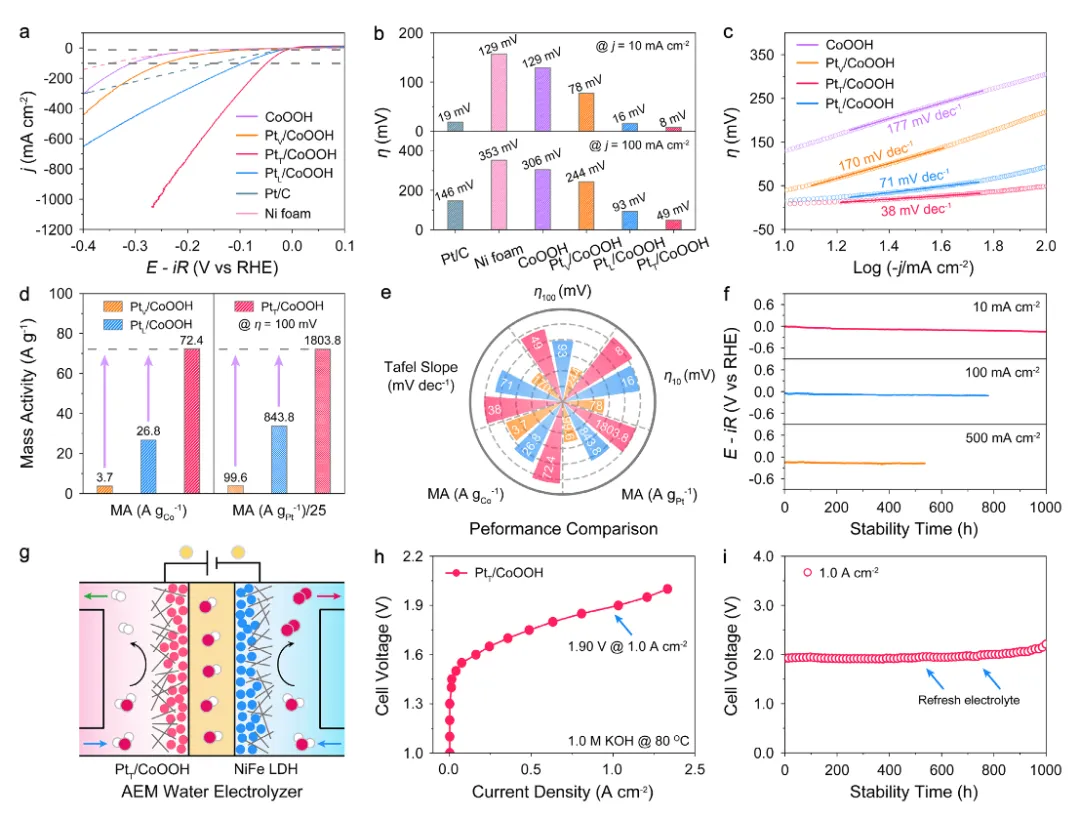
图3:碱性析氢电催化性能
图3全面评估了催化剂的碱性析氢性能。图3a-b展示了极化曲线,PtT/CoOOH在10 mA cm⁻²电流密度下仅需8 mV过电位,比PtL/CoOOH和PtV/CoOOH分别低8 mV和70 mV,在100 mA cm⁻²电流密度下仅需49 mV过电位,比PtL/CoOOH和PtV/CoOOH分别低44 mV和195 mV。PtT/CoOOH仅需255 mV即可达到1000 mA cm⁻²电流密度。图3c展示了Tafel斜率,CoOOH、PtV/CoOOH、PtT/CoOOH和PtL/CoOOH的Tafel斜率分别为177、170、38和71 mV dec⁻¹,PtT/CoOOH的最低Tafel斜率表明其最快的反应动力学。图3d展示了质量活性归一化结果,PtT/CoOOH在100 mV过电位下展现出72.4 A gCo⁻¹,分别是PtV/CoOOH(3.7 A gCo⁻¹)和PtL/CoOOH(26.8 A gCo⁻¹)的19.57倍和2.70倍。PtT/CoOOH的Pt质量活性为1803.8 A gPt⁻¹,分别是PtV/CoOOH(99.6 A gPt⁻¹)和PtL/CoOOH(843.8 A gPt⁻¹)的18.11倍和2.14倍。图3e直观比较了三种催化剂的HER活性,PtT/CoOOH展现出最优性能。图3f展示了长期稳定性测试,PtT/CoOOH在10 mA cm⁻²电流密度下实现了1000小时寿命,在100 mA cm⁻²和500 mA cm⁻²电流密度下分别稳定运行770小时和530小时。图3g-i展示了阴离子交换膜水电解槽(AEMWE)性能,集成PtT/CoOOH的AEMWE仅需1.90 V即可达到1.0 A cm⁻²工业电流密度,并稳定运行1000小时,展现出优异的工业应用潜力。
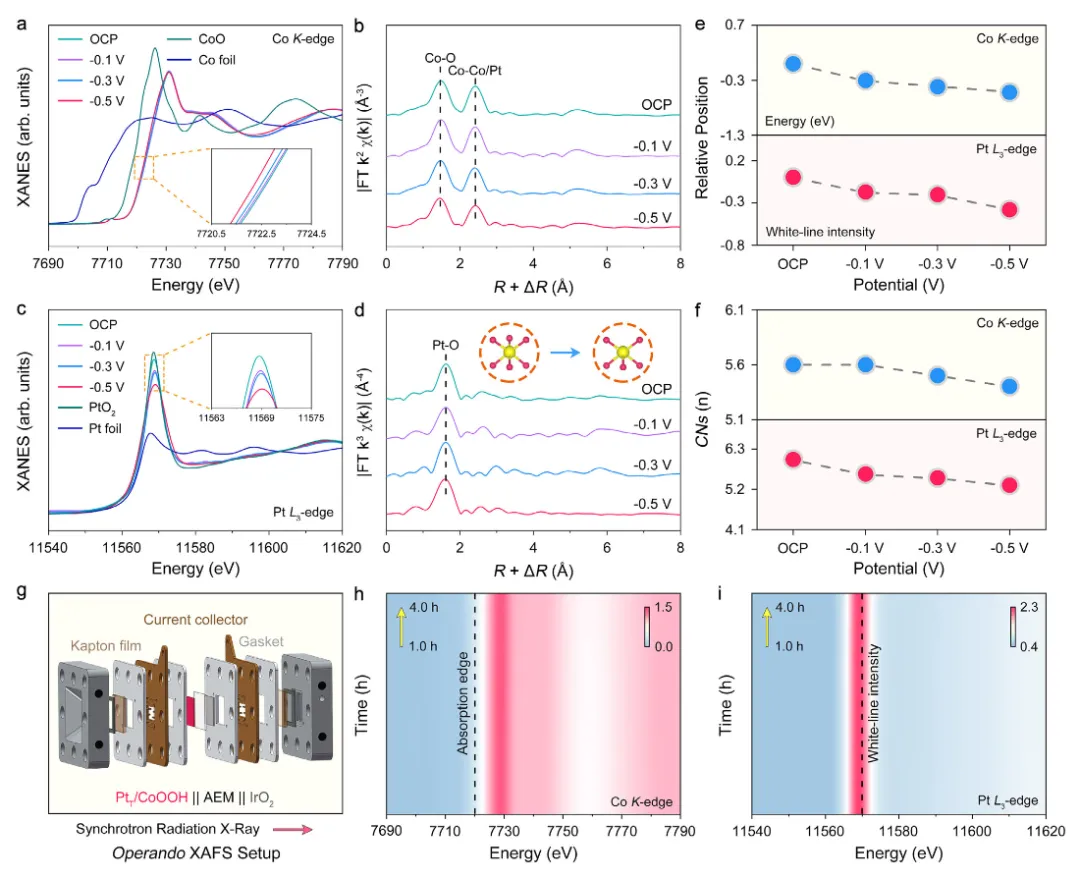
图4:原位/工况XAFS表征Pt和Co物种的动态演变
图4通过原位/工况XAFS技术深入研究了催化剂在HER过程中的动态演变。图4a-f展示了原位Co K边XAFS结果,随着电位从开路电位(OCP)降低到-0.5 V,所有催化剂的Co K边XANES谱图吸收边向低能方向移动,表明Co价态降低。EXAFS拟合结果显示Co-O配位数从初始的约6.0降低到约5.4,表明在HER过程中CoOOH表面发生了部分还原。图4c-e展示了Pt L₃边XANES谱图,随着电位降低,白线峰向低强度方向移动,表明Pt单原子的价态降低。图4d的小波变换分析证实了HER过程中Pt物种保持原子分散状态,未观察到Pt-Pt散射。定量拟合结果显示Pt-O配位数从初始的约6.0降低到约5.3。图4g展示了工况XAFS实验装置示意图,采用透射模式进行测量。图4h-i展示了PtT/CoOOH在HER稳定性测试前后的XAFS对比结果,经过稳定性测试后,Pt L₃边XANES谱图和EXAFS谱图均未发生明显变化,白线强度和Pt-O配位数保持稳定,证实PtT/CoOOH在HER过程中具有优异的结构稳定性。这些原位/工况表征结果揭示了催化剂在HER过程中的动态演变机制,为理解位点依赖的催化性能提供了重要洞察。
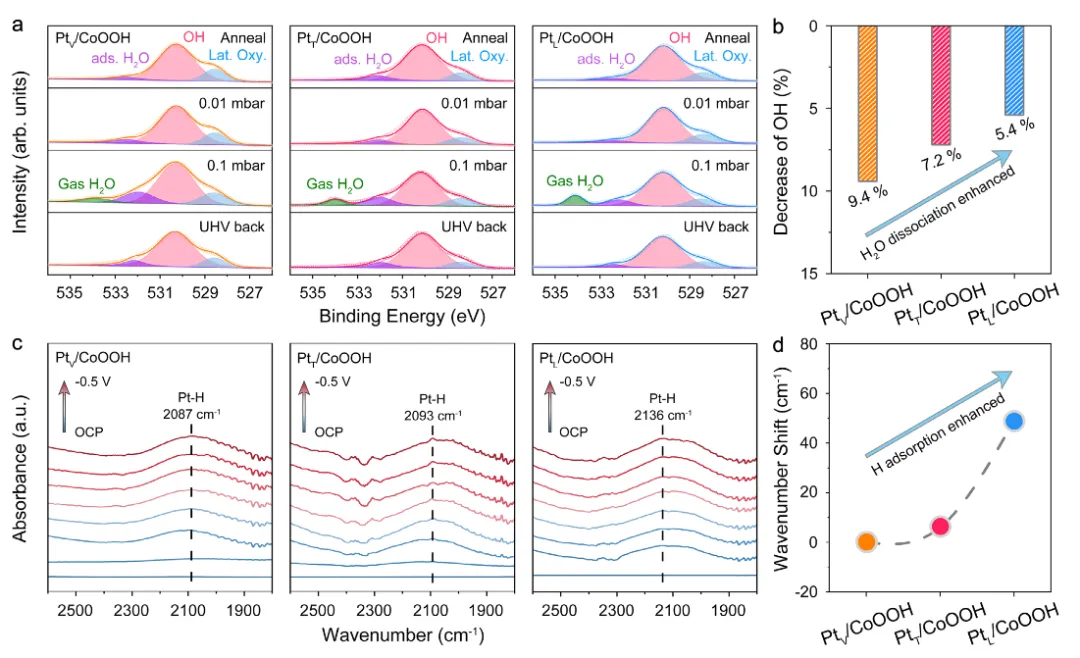
图5:AP-XPS和ATR-SEIRAS表征氢吸附行为
图5通过环境压力X射线光电子能谱(AP-XPS)和衰减全反射表面增强红外吸收光谱(ATR-SEIRAS)深入研究了催化剂的氢吸附行为。图5a展示了Pt 4f XPS谱图的解卷积分析,通过拟合可以将Pt 4f谱图分解为Pt²⁺、Pt³⁺和Pt⁴⁺三个组分。图5b定量分析了三种催化剂中不同价态Pt的比例,PtV/CoOOH、PtT/CoOOH和PtL/CoOOH中Pt⁴⁺的比例分别为2.3%、4.6%和5.4%,这一趋势与XAFS结果一致,证实了从PtV/CoOOH到PtL/CoOOH的Pt价态逐渐降低。图5c展示了ATR-SEIRAS谱图,随着施加电位从OCP降低到-0.1 V,在约2060 cm⁻¹处出现了归属于Pt-H振动的吸收峰,且峰强度逐渐增强,表明在HER过程中Pt位点上发生了氢吸附。图5d展示了三种催化剂在-0.1 V电位下的Pt-H振动峰对比,PtV/CoOOH、PtT/CoOOH和PtL/CoOOH的Pt-H振动峰分别位于2055、2061和2067 cm⁻¹,振动峰向高波数方向移动表明H吸附强度增强。因此,PtV/CoOOH、PtT/CoOOH和PtL/CoOOH展现出依次增强的氢吸附强度。这些结果通过表面敏感的光谱技术直接证实了位点依赖的氢吸附行为,为理解不同位点上Pt单原子的催化性能差异提供了关键证据。
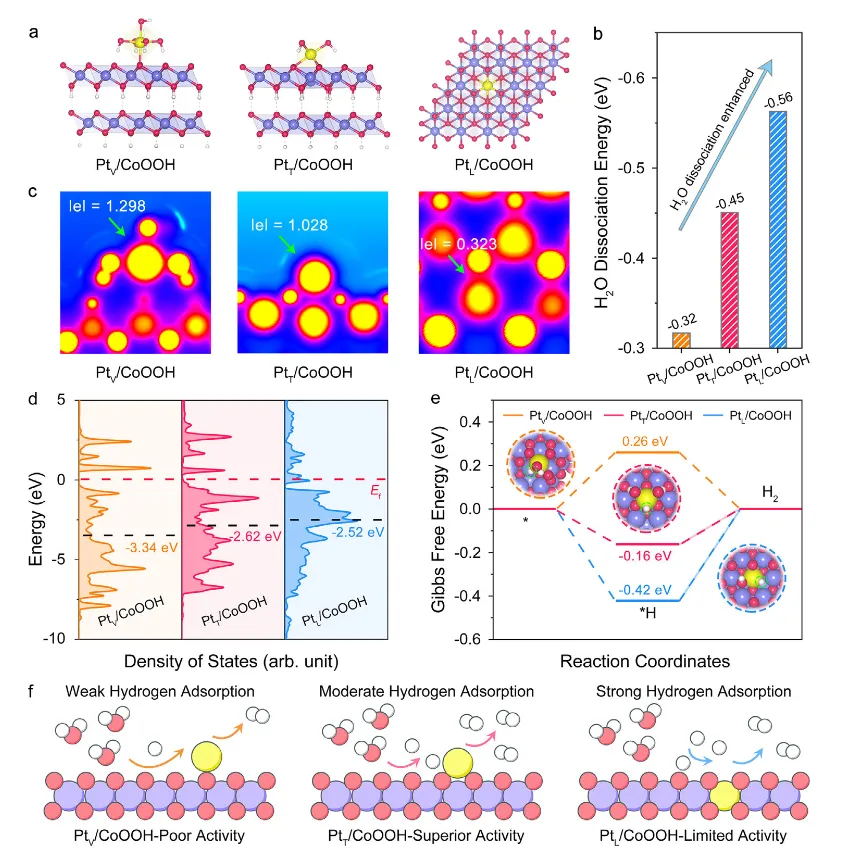
图6:理论计算揭示位点依赖的氢吸附机制
图6通过密度泛函理论(DFT)计算深入揭示了位点依赖的氢吸附机制。图6a展示了理论计算模型,阐明了Pt单原子在不同位点的配位环境。图6b展示了H₂O在Co位点上的解离能,PtV/CoOOH、PtT/CoOOH和PtL/CoOOH的H₂O解离能分别为-0.32、-0.45和-0.56 eV,表明从PtV/CoOOH到PtL/CoOOH,H₂O解离动力学逐渐加快。图6c展示了Pt原子的电子局域函数(ELF)分析,PtV/CoOOH、PtT/CoOOH和PtL/CoOOH的ELF值分别为1.298、1.028和0.323,较低的ELF值对应Pt单原子较低的价态,导致增强的H吸附强度。图6d展示了Pt 5d投影态密度(PDOS),PtV/CoOOH、PtT/CoOOH和PtL/CoOOH的Pt 5d带中心值分别为-3.34、-2.62和-2.52 eV。根据d带中心理论,随着d带中心上移,占据的反键轨道减少,增强了H吸附强度。图6e展示了氢结合能计算结果,PtV/CoOOH、PtT/CoOOH和PtL/CoOOH的ΔG*H-Ads值分别为0.26、-0.16和-0.42 eV,氢结合能降低表明H吸附强度增强。因此,PtT/CoOOH表现出适度的H吸附强度,而PtV/CoOOH和PtL/CoOOH分别表现出最弱和最强的H吸附强度。图6f总结了位点依赖的氢吸附机制:PtV/CoOOH由于缓慢的H₂O解离动力学和最弱的H吸附强度,导致Pt单原子上氢吸附最弱,限制了H₂生成动力学;PtL/CoOOH虽然具有最快的H₂O解离动力学和最强的H吸附强度,但过强的氢吸附导致H₂脱附缓慢和Pt位点中毒;PtT/CoOOH展现出适度的H₂O解离动力学和近热中性的氢结合能,最优的氢吸附促进了平衡的H吸附-H₂脱附动力学,从而实现了优异的碱性HER活性。
总结
本研究通过将Pt单原子精准锚定在CoOOH的三种不同位点(氧空位、三重空位和晶格位点),系统研究了位点依赖的氢吸附行为对碱性析氢反应性能的影响。研究发现,PtT/CoOOH展现出最优的催化性能,在10 mA cm⁻²电流密度下仅需8 mV过电位,并在AEMWE中实现1.0 A cm⁻²工业电流密度和1000小时稳定性。通过原位/工况XAFS、AP-XPS、ATR-SEIRAS和理论计算的综合表征,揭示了PtT/CoOOH具有适度的H₂O解离动力学和近热中性的氢结合能,实现了最优的氢吸附行为。相比之下,PtV/CoOOH因氢吸附过弱限制了H₂生成动力学,而PtL/CoOOH因氢吸附过强导致H₂脱附困难。这项工作不仅开发了精准合成位点特异性单原子的策略,还深入阐明了位点依赖的中间体吸附行为对催化性能的影响机制,为设计高性能电催化剂提供了重要指导。
原文链接
https://doi.org/10.1002/anie.5990615