北京理工大学孙建科/深圳先进院杨新春,最新Angew!离子型有机笼实现界面电荷调控微环境促进电催化硝酸盐还原制氨
- 2026-06-06 08:15:07


第一作者:李书源
通讯作者:孙建科、杨新春
通讯单位:北京理工大学、中国科学院深圳先进技术研究院
氨(NH 3)对农业和工业至关重要,但哈伯-博什工艺能耗高且碳排放量大。电化学硝酸盐还原反应(NO 3 RR)通过将 NH 3 合成与水修复耦合,为农业和工业提供了一种可持续的替代方法。然而,弱 NO 3 吸附能力、竞争性氢进化和亚优化催化剂微环境等挑战阻碍了催化剂的性能。
本研究报告了一系列电催化剂 Pd ⊂QA-Cage x +(x = 24、12、6),它们是通过将 Pd 簇封装在季铵化有机笼中构建而成的。这些离散的宿主可实现均匀的金属团簇封闭和对界面微环境的精确控制。增加笼的电荷密度可丰富界面 NO 3 浓度,移动 Pd d 波带中心,增强 *NO 3 活化。同时,由对离子(Cl -)驱动的电子从 -NH 2 + 转移到笼骨架中生成稳定的自由基,从而介导水活化形成氢自由基(H-),并溢出到 Pd 位点,加速中间氢化。优化的 Pd ⊂QA-Cage 24 + 在中性电解质中的法拉第效率为 95.44%,NH 3 产率为 25.70 mg h -1 mg cat -1,优于电荷较低的类似物。此外,它还能从富营养海水中去除 99.4% 的硝酸盐,降低 NO 3 浓度低于饮用水标准。
要点1. 利用可离子化有机笼作为可编程微环境支架
作者首次将带正电荷的季铵化有机笼(QA-Cage x+)作为模块化、电荷密度可调的宿主,用于封装和调控Pd团簇。通过系统改变笼的电荷密度(从+6到+24),精确地工程化了催化剂-电解质界面的电荷微环境,实现了对反应物(硝酸根)的局部富集和对Pd电子结构(d带中心)的调控,从而协同提升了催化性能。
要点2. 电位触发笼骨架产生长寿命自由基以活化水分子
作者发现,在施加电位时,笼骨架上的季铵基团(-NH2+)可从抗衡阴离子(Cl-)接受电子,生成稳定的氮自由基阳离子。这些长寿命自由基能够高效活化水分子,产生高活性的氢自由基(H•),并通过氢溢流效应供给邻近的Pd活性位点,驱动硝酸根的氢化步骤,有效解决了中性电解质中*H供给不足的难题。
要点3. 构建了集成污染物修复与化学品合成的多功能平台
基于上述设计,最优催化剂Pd⊂QA-Cage 24+在富营养化海水中实现了高效的硝酸盐还原与氨回收。其性能远超现有催化剂,并能将硝酸盐浓度降至饮用水标准以下。这项工作不仅展示了优异的催化性能,更将电催化从单纯的合成拓展至集成式环境修复与资源回收,提供了一个多功能平台的新范式。
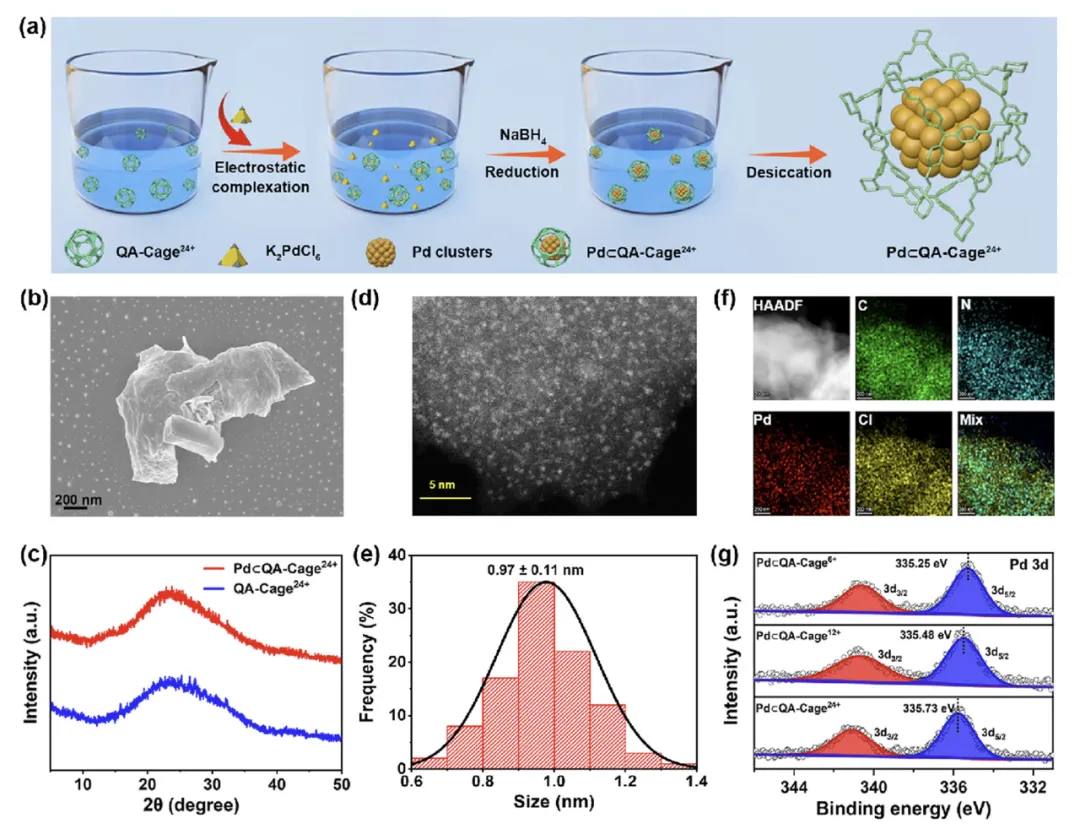
图1 (a) Pd⊂QA-Cageˣ⁺ (x = 24, 12, 6) 合成路线的示意图,以Pd⊂QA-Cage²⁴⁺为例。(b) Pd⊂QA-Cage²⁴⁺的SEM图像。(c) QA-Cage²⁴⁺和Pd⊂QA-Cage²⁴⁺的PXRD图谱。(d) Pd⊂QA-Cage²⁴⁺中Pd簇的HAADF-STEM图像和(e)相应的尺寸分布。(f) Pd⊂QA-Cage²⁴⁺的EDS元素分布图,显示了C(绿色)、N(青色)、Pd(红色)和Cl(黄色)的均匀分布。(g) Pd⊂QA-Cageˣ⁺ (x = 24, 12, 6) 中Pd 3d的XPS谱图。
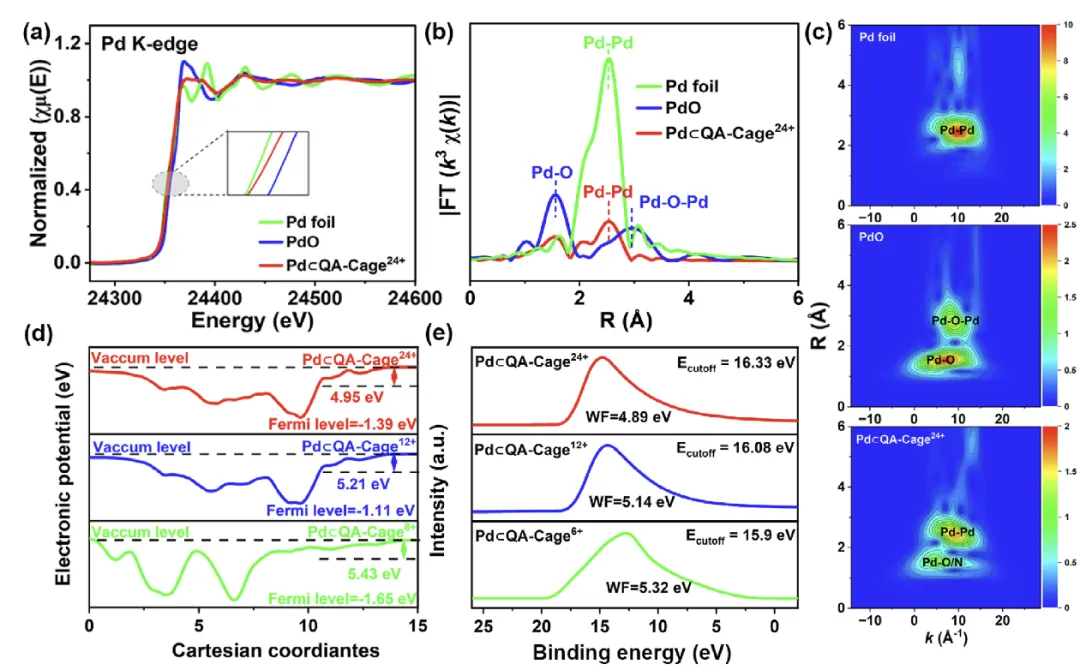
图2 (a) Pd箔、PdO和Pd⊂QA-Cage²⁴⁺的Pd K-edge XANES谱图,(b) k³加权Pd K-edge EXAFS谱图的傅里叶变换,(c) Pd K-edge EXAFS谱图的小波变换。(d) Pd⊂QA-Cageˣ⁺ (x = 24, 12, 6)的功函数计算和(e) UPS谱图。
通过将超小Pd簇封装在可调电荷密度的季铵化有机笼中,成功合成了一系列高性能硝酸根还原电催化剂。其中,优化后的Pd⊂QA-Cage24⁺催化剂在活性上实现了显著提升,其氨产率(25.70 mg h⁻¹ mgcat⁻¹)远超低电荷类似物及商业Pd/C,这主要归因于高电荷笼体通过静电作用富集局部硝酸根浓度,并调控Pd的电子结构(d带中心上移,功函数降低),促进了硝酸根的吸附与活化。在选择性方面,该催化剂实现了95.44%的高氨法拉第效率,并大幅抑制了亚硝酸根副产物和析氢副反应,这得益于笼体骨架在电位触发下产生的氢自由基(H•)有效促进了氢化步骤,以及热力学上对析氢的抑制。在稳定性方面,催化剂在长期运行和循环测试中保持了结构和性能的稳健。此外,该催化剂展现出优异的实际应用潜力,在成分复杂的真实富营养化海水中仍能高效工作,实现了超过99.4%的硝酸盐去除和氨回收,展示了其在集成污染物修复与可持续合成中的价值。
文献链接:
https://doi.org/10.1002/anie.4155022
以上内容,如有误读和纰漏,敬请指正
解决世纪争议难题,郑州大学“最硬核团队”重磅Nature: 合成纯相块状六方型金刚石 院士携手!兰州大学,校史首篇Nature Nanotechnology:飞秒时间尺度上的质子 - 电子时间异步性,实现质子交换膜电解槽用耐腐蚀低铱阳极 分子筛催化,再发Nature Catalysis! 南开大学陈军院士,2025年成果汇总 最新JACS,高稳定负载型 Cu-Ni 稀释合金催化剂实现高效 CO2 重整甲烷 中科大,最新Nature Chemistry!双原子催化剂! 杰青联手!华东师范大学关小红/孙远奎&湖南大学王双印,最新Angew:硫掺杂零价铁气凝胶*NO偶联和*H供给协同调控驱动硝酸盐高效电还原成N2 湖南大学「国家杰青」王双印&中南大学任博华&天津大学杨娜,最新Angew:机器学习揭示有机-金属界面“电子海绵”行为促进CO2电还原C2+生成 李灿院士领衔!兰州大学李泽龙,最新Angew:NdO强化IrMnOx双位点协同,酸性OER低Ir长寿命! 韩布兴院士领衔!中科院化学所张裴,最新Angew!温度介导动力学与界面控制以实现浓缩热敏性生物质原料持续电氧化!

如需转载或合作,请联系我们
联系方式:15715750735(微信同)
联系邮箱:mon@xueyanhui.com
2.催化进展现有综合群、电催化交流群、同步辐射交流群、文献交流互助群、各研究领域群等近20余个,欢迎大家加小编微信,我们会尽快拉您进入对应的群。


