深圳大学骆静利/刘绍庆最新AFM丨调控水取向实现安培级CO!
- 2026-04-25 07:41:53

电化学CO2还原制甲酸盐是可持续化工生产路径,但高电流密度下质子供给不足一直是工业化应用的核心瓶颈。传统研究多聚焦催化剂电子结构调控与CO2活化,界面水构型对水解离动力学的调控作用长期被学界忽视。
近年来,在析氢等反应中研究发现,氢向下水构型的解离能垒远低于氧向下水,可加速质子生成,为CO2还原催化提供了新思路。
2026年3月18日,深圳大学骆静利、刘绍庆、Shu-Wen Wu团队在Advanced Functional Materials发表了标题为《Engineering Hydrogen-Down Water Configurations for Ampere-Level Electrochemical CO2 Reduction to Formate》的论文,Xiao-Dong Guo为论文第一作者,骆静利、刘绍庆、Shu-Wen Wu为论文共同通讯作者。

论文亮点: 1. 首次在CO2还原体系中实现界面水取向的微观调控; 2. 利用HfO2作为分子开关,诱导水从氧向下转变为氢向下取向; 3. 氢向下水构型显著降低水解离与CO2加氢能垒; 4. 实现安培级电流密度下97%的甲酸盐法拉第效率; 5. 膜电极组装器件稳定运行超过60 h,满足工业应用要求。
📄 全文速览
电化学CO2还原制甲酸盐是一条可持续的化工生产路径,但高电流密度下质子可及性有限,阻碍了工业化应用。以往研究多聚焦催化剂电子结构调控与CO2活化,界面水构型调控水解离动力学的作用一直被忽视。
作者利用HfO2作为铋表面的分子开关,调控界面水分子取向,更倾向于形成氢向下而非氧向下构型。从头算分子动力学模拟和密度泛函理论计算证实,氢向下构型大幅降低水解离能垒,产生活性氢物种,多种原位表征验证了界面水结构变化。
HfO2修饰铋实现了优异的甲酸盐分电流密度(-970 mA cm-2),法拉第效率达97%。
在膜电极组装器件中,催化剂在0.2-1.6 A范围内均保持90%以上的甲酸盐选择性,证明界面水工程是工业化CO2还原催化剂开发的有效新策略。
📊 图文解读
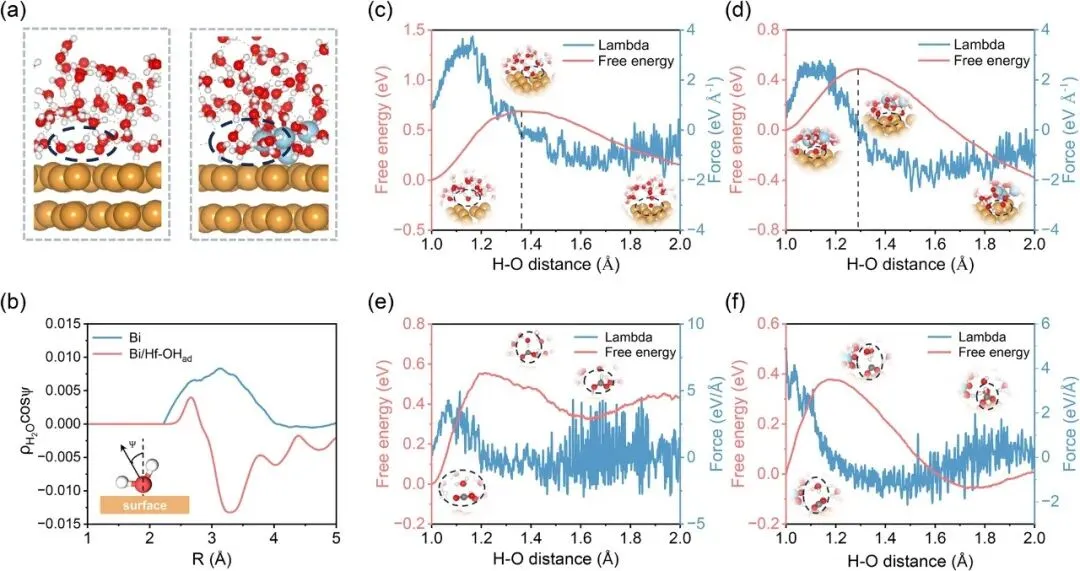
图1 | (a) Bi表面和Bi/Hf-OHad表面动态取向水的界面结构代表性快照;(b) 沿距离方向,Bi上方和Bi/Hf-OHad上方界面水的偶极取向(ρH2O cos ψ),经10 ps计算平均得到;水解离过程的自由能随反应坐标CV的变化,分别在(c) Bi和(d) Bi/Hf-OHad表面;CO2加氢过程的自由能随反应坐标CV的变化,分别在(e) Bi和(f) Bi/Hf-OHad表面
模拟结果表明,原始Bi表面的界面水分子主要采取氧向下取向,而Bi/Hf-OHad表面通过Hf-OH与水分子的氢键作用,驱动水分子重新取向为氢向下构型,偶极矩方向反转证明了该结构转变。
水解离活化能从原始Bi表面的0.69 eV大幅降至Bi/Hf-OHad表面的0.48 eV,CO2加氢活化能也从0.56 eV降至0.37 eV,证实氢向下水取向可促进质子转移与CO2加氢过程。
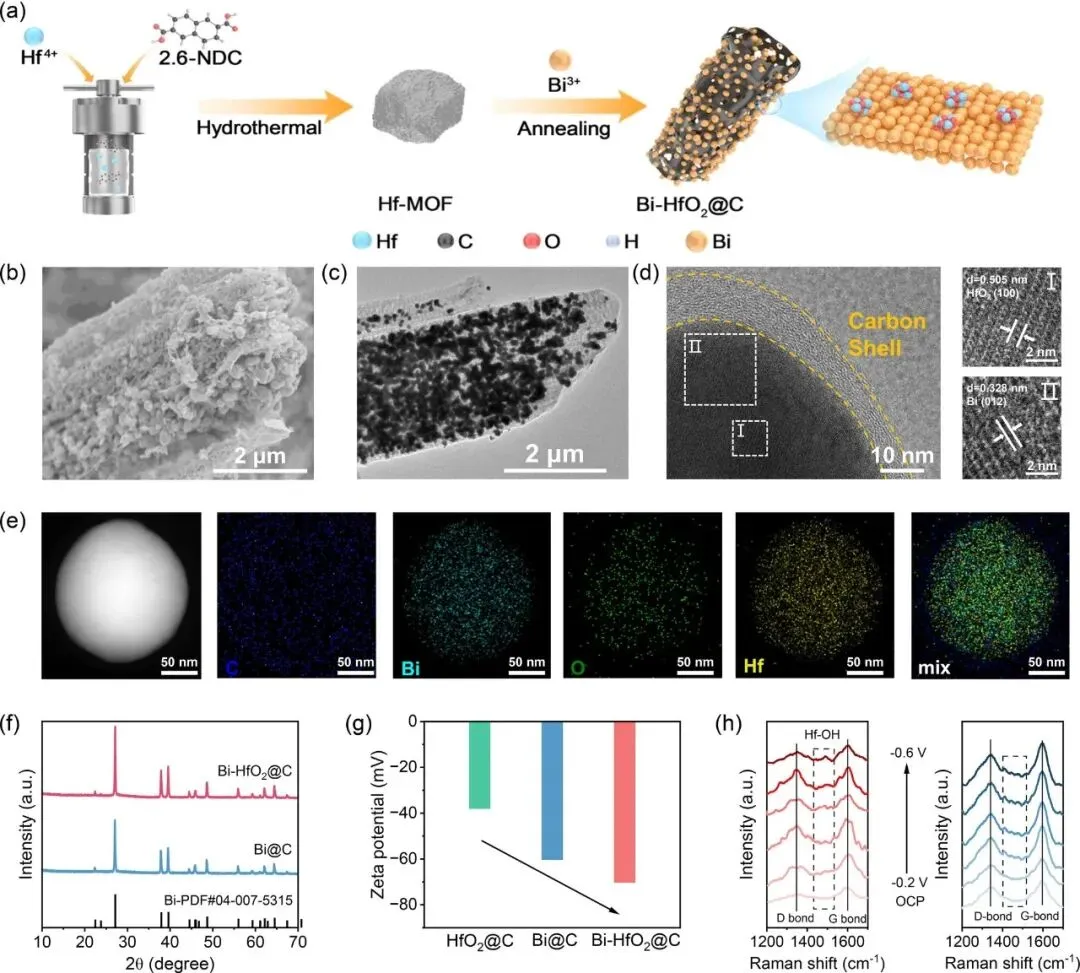
图2 | (a) Bi-HfO2@C合成流程示意图;(b) Bi-HfO2@C的SEM图像;(c) Bi-HfO2@C的TEM图像;(d) Bi-HfO2@C的HRTEM图像;(e) 对应的EDX元素 mapping;(f) Bi-HfO2@C和Bi@C的XRD图谱;(g) Bi-HfO2@C、Bi@C和HfO2@C的Zeta电位值;(h) ECR过程不同电位下Bi-HfO2@C和Bi@C的原位拉曼光谱
作者通过两步法成功合成了多孔碳负载的HfO2修饰Bi催化剂Bi-HfO2@C,表征结果显示,约200 nm的纳米颗粒均匀分散在多孔碳基体中,可检测到对应HfO2(100)晶面和Bi(012)晶面的晶格条纹,证明了复合材料的成功制备。
原位拉曼光谱证实,电化学还原条件下催化剂表面可形成稳定吸附羟基Hf-OHad,为调控水取向提供结构基础。
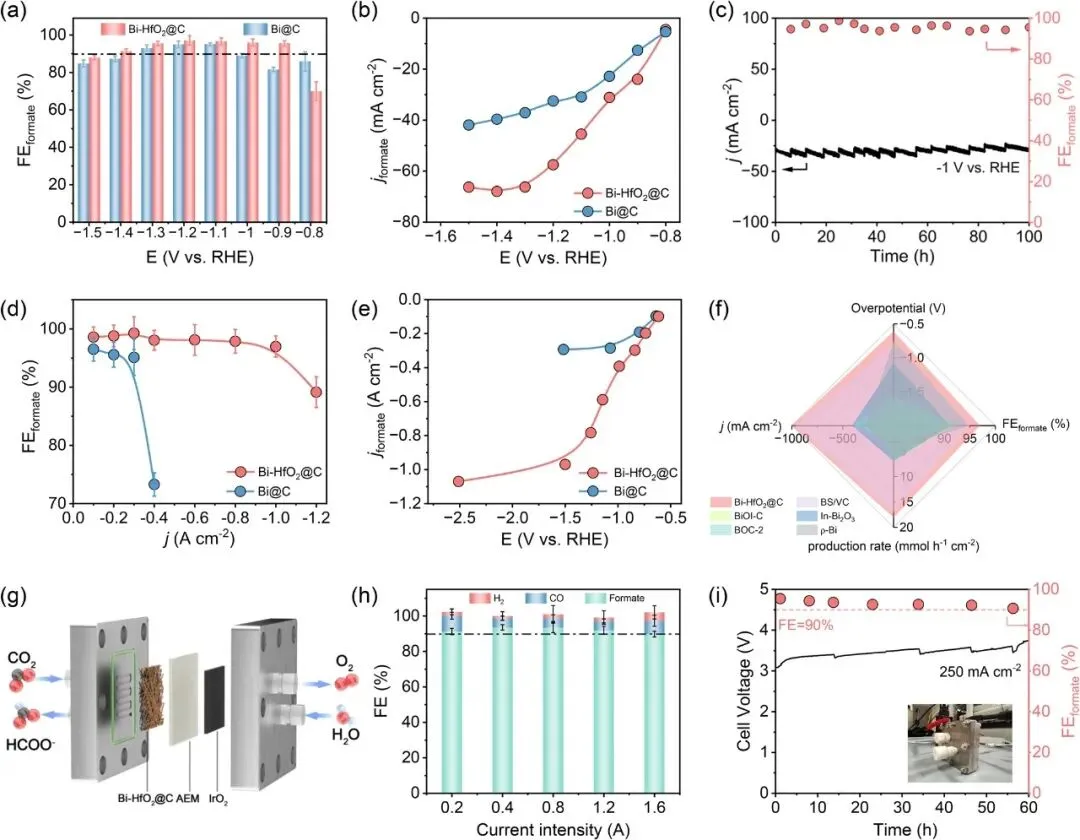
图3 | (a) 在H型电解池CO2饱和0.5 M KHCO3溶液中Bi-HfO2@C和Bi@C的甲酸盐法拉第效率;(b) 上述体系中Bi-HfO2@C和Bi@C的甲酸盐分电流密度;(c) Bi-HfO2@C在-1 V下的稳定性测试;(d) 在流动电解池1 M KOH溶液中Bi-HfO2@C和Bi@C的甲酸盐法拉第效率;(e) 上述体系中Bi-HfO2@C和Bi@C的甲酸盐分电流密度;(f) Bi-HfO2@C与近期已报道铋基催化剂的电流、过电位(200 mA cm-2下)、甲酸盐法拉第效率和产率对比;(g) 采用阴离子交换膜电解槽的MEA结构示意图(2×2 cm2);(h) MEA电解槽中1 M KOH溶液不同施加电流下的法拉第效率;(i) Bi-HfO2@C在MEA电解槽中的长期稳定性测试
电化学性能测试表明,Bi-HfO2@C在H型电解池中-0.9至-1.4 V宽电位窗口内甲酸盐法拉第效率保持90%以上,峰值达97.1%,-1.4 V下甲酸盐局部电流密度达-61.8 mA cm-2,且可稳定运行100 h。
在流动电解池中,该催化剂在-1 A cm-2下仍保持97%以上的甲酸盐法拉第效率,最大甲酸盐局部电流密度达-1.069 A cm-2。
在4 cm2膜电极组装器件中,催化剂在0.2-1.6 A宽电流范围内保持90%以上甲酸盐选择性,1 A恒电流下可稳定运行超过60 h,具备工业化应用潜力。
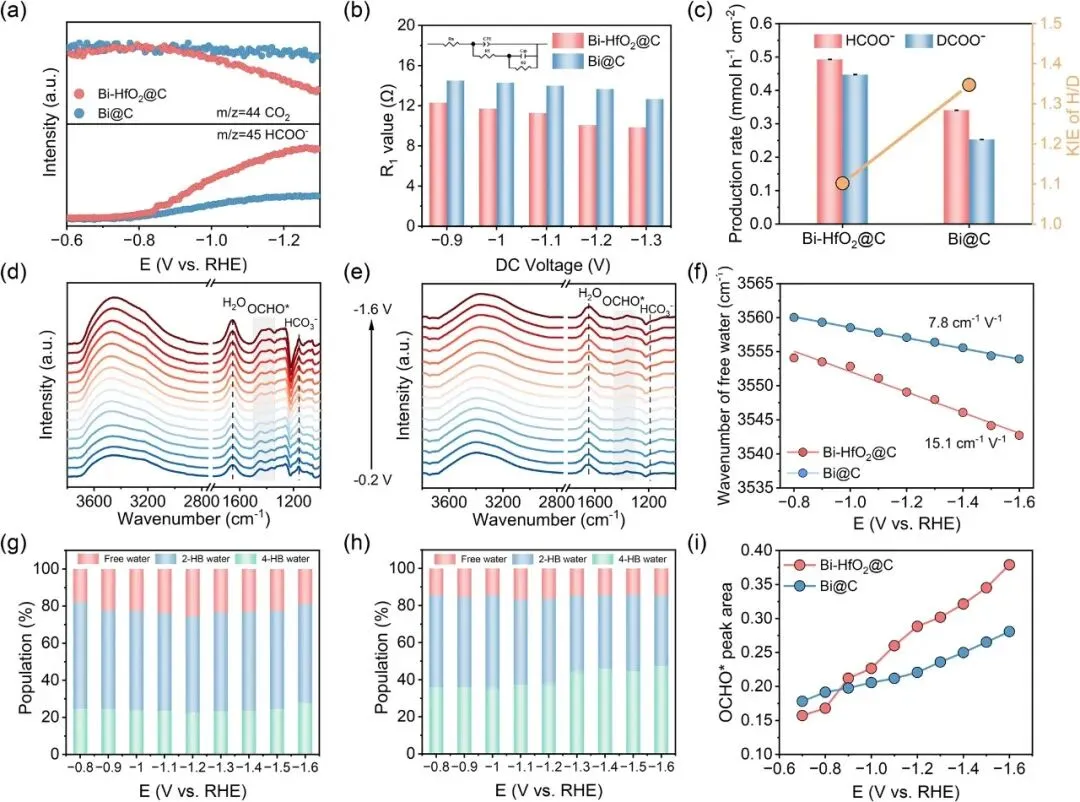
图4 | (a) ECR制甲酸盐过程中Bi-HfO2@C和Bi@C的在线DEMS分析;(b) 由Nyquist图推导得到,不同直流电压下Bi-HfO2@C与Bi@C在1 M Ar饱和KOH中的R1值对比;(c) -1.1 V vs RHE下由甲酸盐生成速率推导得到Bi-HfO2@C和Bi@C的H/D动力学同位素效应;不同电位下在CO2饱和0.5 M KHCO3溶液中记录的原位ATR-SEIRAS光谱,分别在(d) Bi-HfO2@C和(e) Bi@C表面;(f) Bi-HfO2@C和Bi@C表面自由水的O-H伸缩振动斯塔克调谐率对比;不同电位下界面水比例变化,分别在(g) Bi-HfO2@C和(h) Bi@C表面;(i) 不同电位下Bi-HfO2@C和Bi@C的OCHO*峰面积对比
机理研究证实,Bi-HfO2@C的水活化电荷转移电阻更低,动力学同位素效应数值更小,证明水解离动力学更优异。
原位红外光谱结果显示,Bi-HfO2@C表面自由水比例更高,自由水的斯塔克调谐率更大,证实氢向下水构型的存在,同时关键中间体OCHO*的信号更强,证明该构型可促进水解离、加速质子供给,从而提升CO2加氢效率。对比不同氧化物修饰催化剂,性能与自由水含量正相关,进一步验证了该机制。
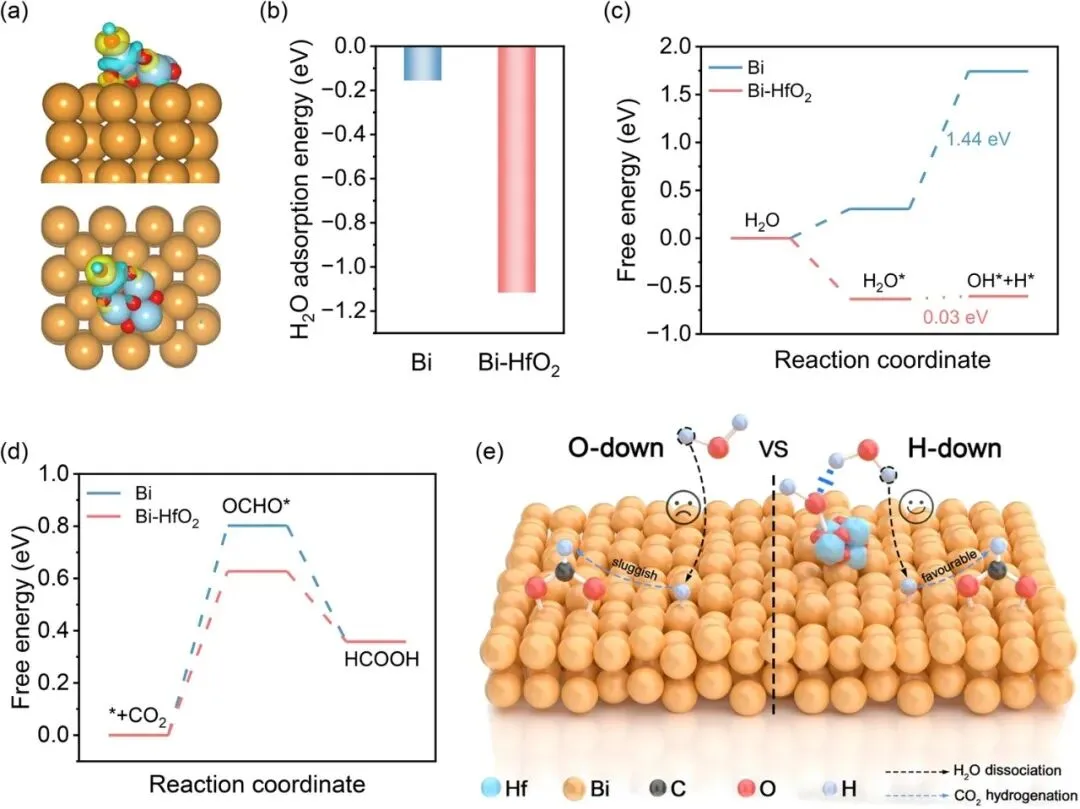
图5 | (a) 不同视角下吸附OH的Bi-HfO2的电荷密度差分图;(b) Bi和Bi-HfO2表面的H2O吸附能;(c) Bi-HfO2和Bi表面水解离过程的吉布斯自由能图;(d) Bi-HfO2和Bi表面甲酸生产的吉布斯自由能图;(e) Bi-HfO2表面氢向下水解离以及Bi表面氧向下水解离的示意图
密度泛函理论(DFT)计算结果显示,HfO2修饰后,Hf相邻位点存在局域电荷积累,对水分子的吸附能更强,水解离步骤的吉布斯自由能变从原始Bi的1.44 eV大幅降至Bi-HfO2的0.03 eV,OCHO*形成能垒也从0.802 eV降至0.627 eV,证实Hf-OHad诱导的氢向下水取向可促进水活化、加速H*生成,从而降低CO2加氢能垒,从理论层面验证了实验与原位表征结果。
📝 总结
综上,作者报道了通过微观水取向工程实现工业级电化学CO2还原制甲酸盐电解。
从头算分子动力学模拟、密度泛函理论计算和原位表面增强红外吸收光谱测试均证明,Bi表面修饰HfO2可引导界面水采取更有利的氢向下取向,而非传统的氧向下取向。
该结构使Volmer步骤活化能显著降低,确保高电位下可持续提供充足氢,驱动CO2加氢过程。
最终Bi-HfO2催化剂在1 A cm-2电流密度下实现97%的高甲酸盐法拉第效率,性能优于目前已报道的大多数铋基催化剂。
在膜电极组装器件中,该催化剂在0.2-1.6 A宽电流范围内均保持90%以上的甲酸盐选择性,可稳定运行超过60 h。本研究突出了界面水取向对实现工业化CO2电解的关键作用,该策略有望推广至其他涉及加氢过程的催化反应。
【文献信息】 Engineering Hydrogen-Down Water Configurations for Ampere-Level Electrochemical CO2 Reduction to Formate. Adv. Funct. Mater., 2026, https://doi.org/10.1002/adfm.74953.
#深圳大学#骆静利#刘绍庆#催化剂#AFM#甲酸盐电解#法拉第效率


