背靠背连读 | 深圳湾实验室冯根生教授现象级巨作,揭秘癌细胞耐药的新型细胞通讯方式
- 2026-06-14 05:24:31
在科研内卷到极致的今天,很多基础与转化研究陷入了一个套路化的怪圈:敲除个基因/加个药 + 跑个多组学测序(单细胞/空间) + 凑几条差异通路 = 一篇SCI。
这种流水线式的数据堆砌,看似图表华丽,实则缺乏灵魂。审稿人和工业界最常问的一句话往往是,你的表型和信号通路变化确实存在,但驱动这一切的物理和分子底层机制(Underlying Mechanism)到底是什么?
真正的顶级研究,绝不是大量数据堆砌,而是以强大的逻辑为刃,剖开复杂生命现象的内核。
近期,加州大学圣地亚哥分校(UCSD)冯根生教授团队 (现全职加入深圳湾实验室,任肿瘤研究所资深研究员、所长)的两项连续性重磅研究,分别为我们提供了一个教科书级的原创性研究案例。
一篇发表于顶刊《eLife》(2023),题为《Identification of CD133+ intercellsomes in intercellular communication to offset intracellular signal deficit》。


这两篇文章虽未首发于 CNS,但其从 0 到 1 发现新型细胞通讯方式的原创性与潜力被远远低估。如果不看影响因子(IF),只看颠覆因子(D-index),其科研价值远超许多常规的修补型 CNS 文章。

在评价科研成果时,唯 IF 论是一个极大误区。
🔹 影响因子(IF):衡量期刊的短期流量。反映过去两年的平均被引频次,极易被热门领域的跟风研究推高,掩盖了冷门期刊中的开创性工作。
🔹 颠覆因子(D-index):精准过滤水文,锁定诺奖级原创。它通过引文网络算法考察研究是否改变了科学轨迹。如果后人引用你的论文时,不再引用你的参考文献,说明你实现了从 0 到 1的颠覆;若后人依然大量引用你的前人,说明你只是在前人的树干上修补。
今天,【药研逻辑】将带您背靠背硬核拆解这两篇现象级巨作。看顶尖团队是如何抽丝剥茧,完成从“现象发现 ➡️ 结构鉴定 ➡️ 机制破解 ➡️ 联合用药”的完美转化闭环的。
💥 上篇:打破常规,揪出隐秘的全新细胞器细胞间体(intercellsome)
Identification of CD133 intercellsomes in intercellular communication to offset intracellular signal deficit. eLife. 2023
寻找决定细胞命运的开关,最需要的是对反常现象的敏锐度。传统的科研论文往往只关注常规的差异基因,但冯教授团队没有因为反常的数据而草草替换方法或者研究方向,而是紧抓活体动物中的反常表型思考,最后成功提出颠覆性的研究。
常规思维中,当促进增殖的核心信号分子(如 Shp2)被敲除后,肝脏再生理应全面停滞。
之前的文章我们介绍了SHP2 悖论。(点击回顾:从 SHP2 悖论看实体瘤靶向治疗的生态学转向):
在肝脏中,SHP2 根本不是促癌恶人,而是维持稳态的肿瘤抑制因子。强行单点敲除它,会引发极其严重的肝脏炎症、坏死与代偿性增生、衰老以及免疫抑制微环境。
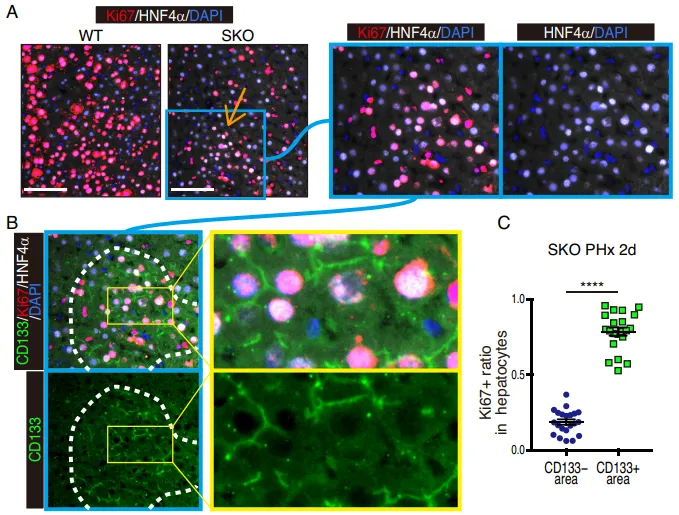
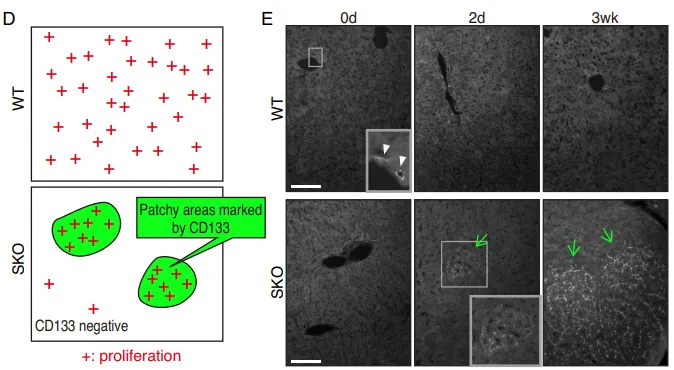
Fig. 1. 鉴定部分肝切除(PHx)后 Shp2 缺陷(SKO)肝脏中斑片状的肝细胞增殖模式
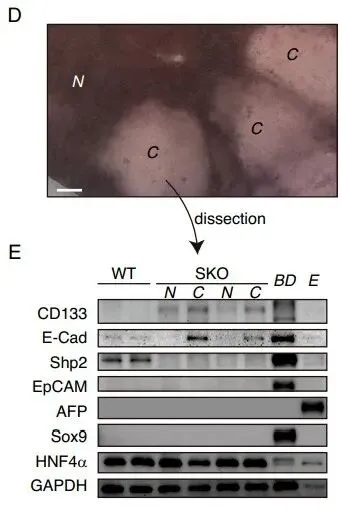
Fig. 2. CD133+ 细胞集落代表 Shp2 缺陷肝脏中独特的再生过程
但在真实的活体小鼠中,团队利用免疫荧光(IF)发现。在 Shp2 敲除(SKO)的小鼠切除部分肝脏后,并非所有肝细胞都停止了分裂。部分异质性的肝细胞聚集成簇,强行开启了增殖(Fig. 1A-1D)。通过严苛的标志物排查,他们发现这些顽强增殖的细胞簇高度特异性地表达 CD133(Fig. 1E),但并不表达其他常见的祖细胞以及干细胞标志物(如 EpCAM、AFP、Sox9)(Fig. 2D-2E)。
PS:亮点(打破 CD133 刻板印象):这直接打破了CD133 仅仅是肿瘤干细胞(Cancer Stem Cell,CSC)静态标志物的固有认知,证实这些抱团细胞是成熟肝细胞在面临信号匮乏时的应急状态,而非简单的干细胞扩增。
如果这种现象只存在于肝切除模型中,其产业价值将大打折扣。
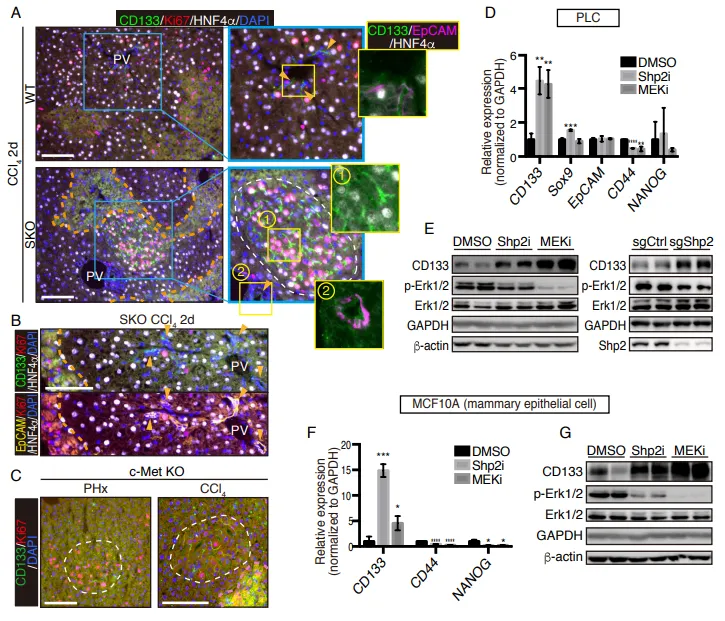
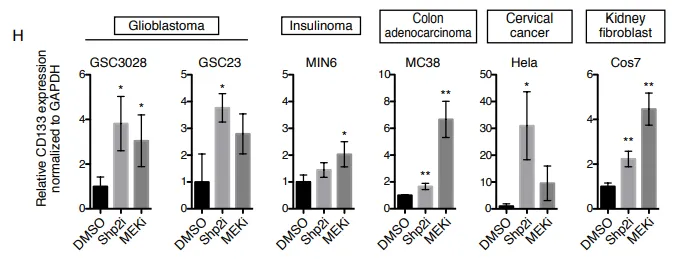
PS:亮点(跨物种与泛癌种的普适性铁证):无论正常细胞还是癌细胞,只要面临“增殖信号受损”的绝境,都会本能地上调 CD133。这证明其不是特例,而是一种高度保守的泛癌种应激求生底层机制。
这些高表达 CD133 的抱团细胞到底在交流什么?传统的细胞通讯分析,如 cellchat、cellphonedb 及 linna 等生信工具只能列出一堆配受体对。但团队直接上了物理铁证。
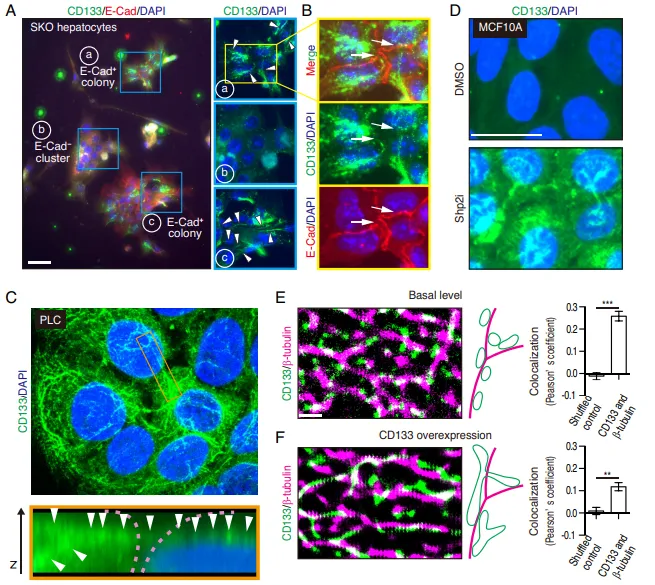
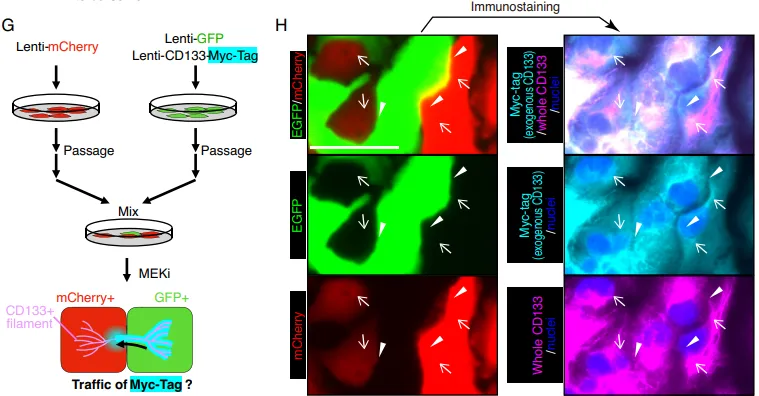
原代细胞的跨细胞连线。观察体外培养的 SKO 原代肝细胞时发现,CD133 并没有像普通膜蛋白那样均匀分布在细胞表面,而是形成了一条条丝状结构(Fig. 5A)。在 E-Cad 阳性的抱团细胞群中,这些丝状物直接跨越细胞边界,像缆绳一样把相邻细胞死死连在一起(Fig. 5B)。
3D 共聚焦立体实锤。为证明这绝非二维错觉,团队利用 3D 重构对肝癌细胞(PLC)进行扫描。Z 轴切面极其直观地展示,CD133 形成了一条连续的空中走廊,架设在两个相邻细胞之间(Fig. 5C)。
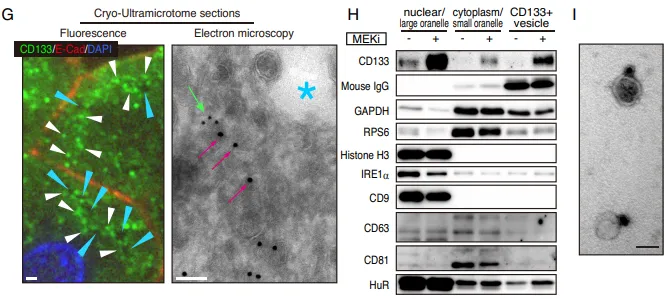
排雷与结构实锤。团队使用不同种属抗体排除假阳性(Fig. 5D)。为了搞清这究竟是什么材质,团队动用了显微成像的核武器,超分辨显微镜(STORM)和免疫电镜(EM)。结果证实,这些看似连续的丝线,实际上是聚集在微管纤维网络上的、直径约 50 nm 的微小囊泡(Fig. 5E-5G),且不含经典外泌体标志物(Fig. 5H)。团队正式将其命名为全新细胞器,细胞间体(Intercellsome)。
这些新型非经典囊泡 Interellsome 里到底装了什么?
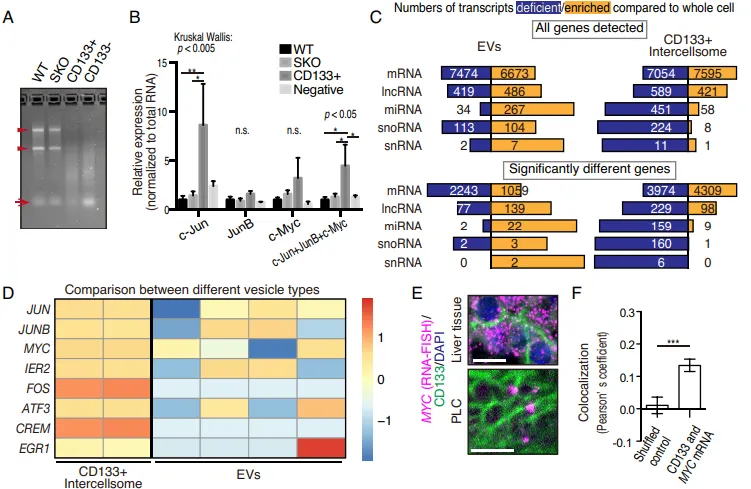
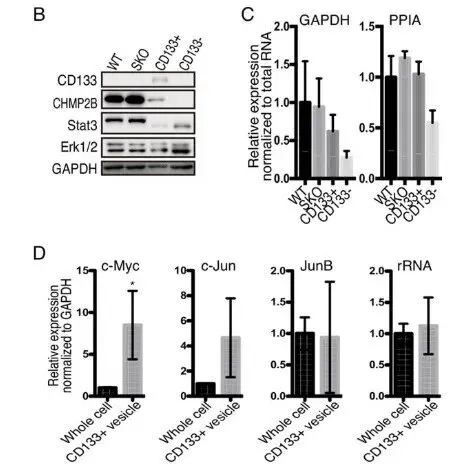
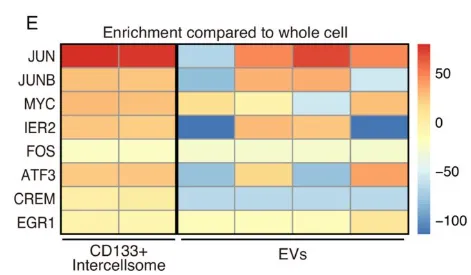
纯化出的 CD133 细胞间体与普通外泌体(EVs)截然不同。普通 EVs 富集 micro-RNAs,而 Intercellsome 极度富集信使 RNA(mRNAs)(Fig. 6A-6C)。单分子 RNA-FISH 更是直接拍到了 Myc、Jun 等早期促增殖响应基因(IEGs)的转录本正搭载着 CD133 囊泡在细胞间穿梭(Fig. 6E)。qPCR 进一步证实,这些囊泡精准装载了生存所需的战略物资(Supp Fig 1B-1E)。
PS:亮点(超分辨成像与货物的物理实锤)。团队没有依赖传统的生信推测,而是直接利用超分辨电镜提供了实体物理桥梁的视觉铁证。同时,证明其装载的是 mRNA 而非 miRNA,彻底确立了新细胞器的独立身份。

🔹 经典外泌体 (Exosome / EVs):专属身份证为 CD63+、CD9+、CD81+。由细胞内多囊泡体释放,在细胞外空间自由漂浮,像漂流瓶一样远距离递送 micro-RNA 和蛋白,进行常规的系统性调节。
🔹 新型细胞间体 (Intercellsome):专属身份证为 CD133+。由细胞质膜直接出芽,沿着相邻细胞间的实体丝状纤维定向滑行。像实体桥梁一样递送巨大的完整信使 RNA (mRNAs),核心使命是在绝境中代偿受损的增殖信号。

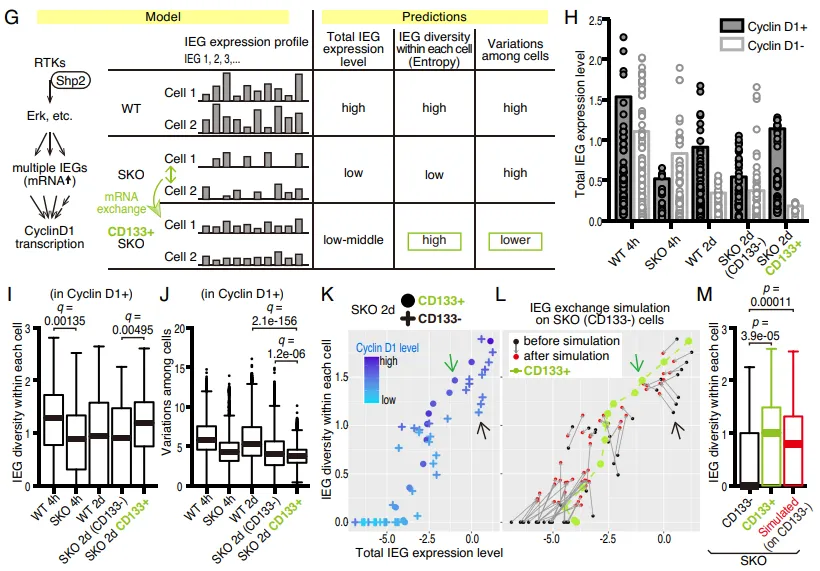
单细胞 RNA 测序(scRNA-seq)揭示,在 Shp2 缺失的细胞中,促分裂 mRNA 的细胞内多样性(即熵值,entropy)极低,且充满随机性。但数学建模与测序数据拟合后证实:当这些有缺陷的细胞通过 Intercellsome 互相交换残存的 mRNA 时,它们成功将细胞间的随机异质性转化为了细胞内部的基因多样性(熵增)(Fig. 7I-7M)。
PS:亮点(数学建模与单细胞组学的完美结合):不玩常规的单细胞分析套路,团队首次引入熵(Entropy)概念,用精妙的数学推演完美解释了细胞如何通过共享资源实现群体求生,展现了极高的计算生物学品味。
五、 活体绝杀:彻底切断退路
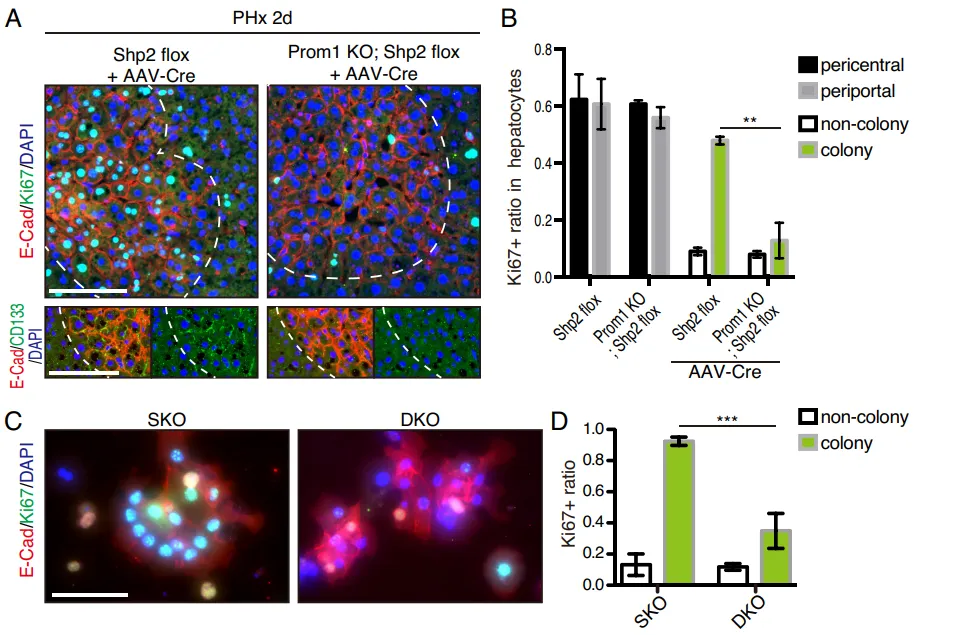
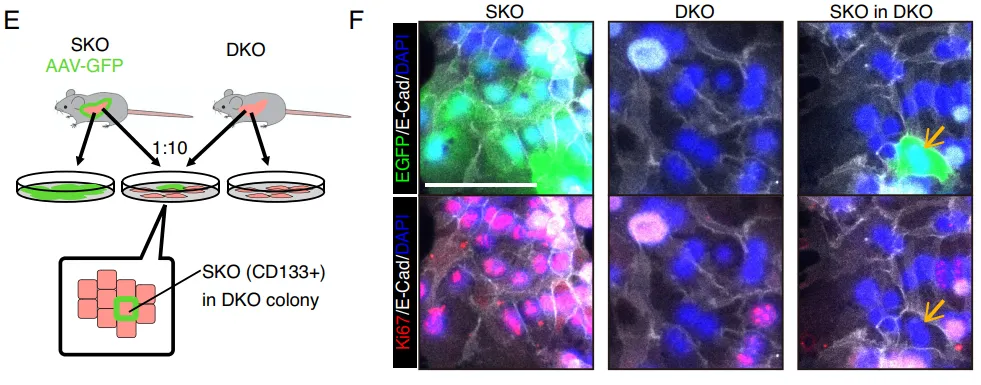
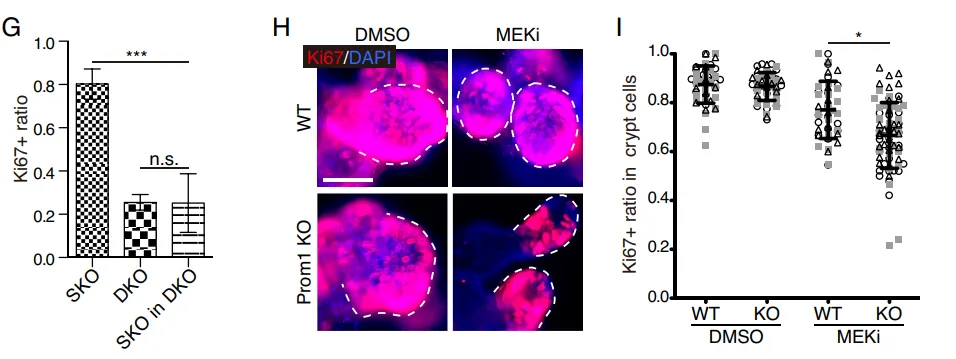
团队繁育了 Prom1 (CD133) 与 Shp2 的双敲除(DKO)小鼠。结果显示,一旦在体内剥夺了 CD133,那些原本能够代偿增殖的细胞簇瞬间大幅失去了增殖能力(Fig. 8A-8B)。在体外肠道类器官中,缺失 CD133 的类器官面对 MEK 抑制剂的打击时,增殖彻底停滞(Fig. 8H-8I)。
PS:亮点(活体层面的基因双敲绝杀)。在活体小动物模型中直接剥夺 CD133 基因,导致代偿增殖瞬间崩溃,无可辩驳地坐实了 Intercellsome 作为抗压救生圈的核心生命功能。
🎯 下篇:直击药研痛点,揭秘 YAP 如何操纵新型通讯网络打破耐药
CD133+ Vesicles Mediate Resistance to RAS-ERK Inhibition Regulated by YAP Activation in Liver Cancer Cells. bioRxiv. 2025
上篇在生理模型中发现了 Intercellsome。但这留下了一个巨大的产业悬念:在真实的抗肿瘤靶向药(如 MEK 抑制剂)压力下,这条地下暗网真的起作用吗?是谁在幕后给细胞下达了开启 Intercellsome 的指令?
2025 年的最新研究,直接将场景切换到了工业界最头疼的痛点,靶向药耐药。团队通过极致的实验设计,把审稿人和工业界可能提出的质疑全部堵死。
一、 现象的泛化,这不是特例,是靶向药逼出的求生本能
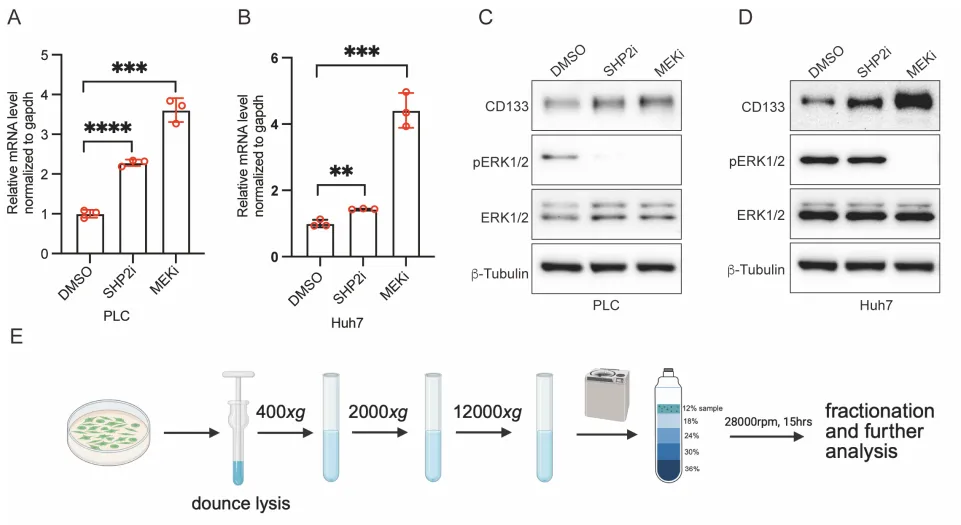
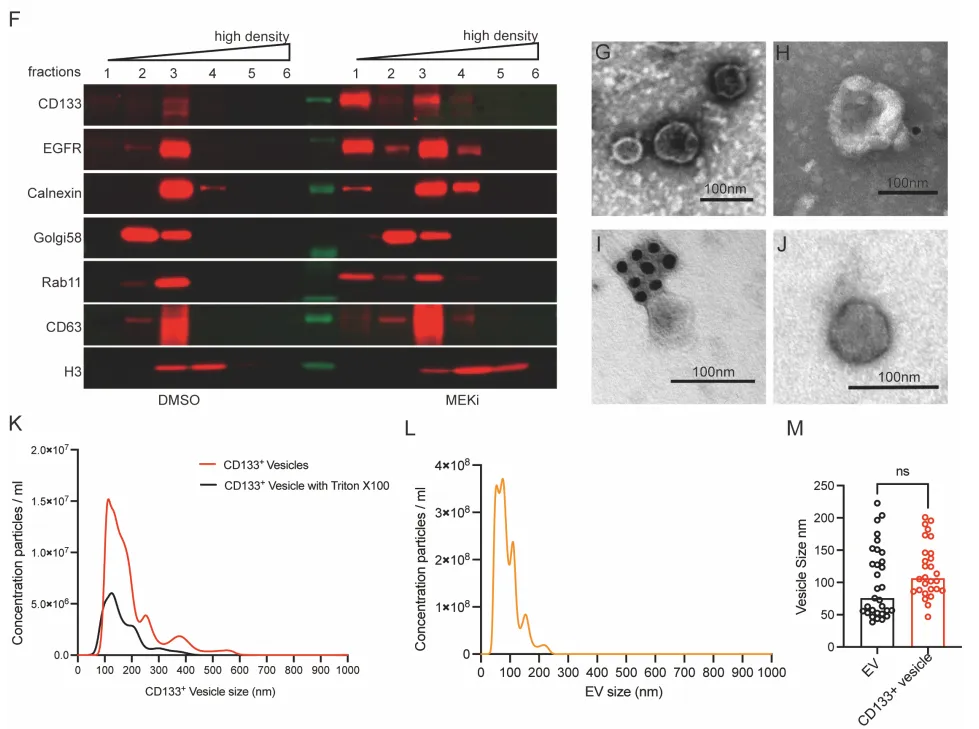
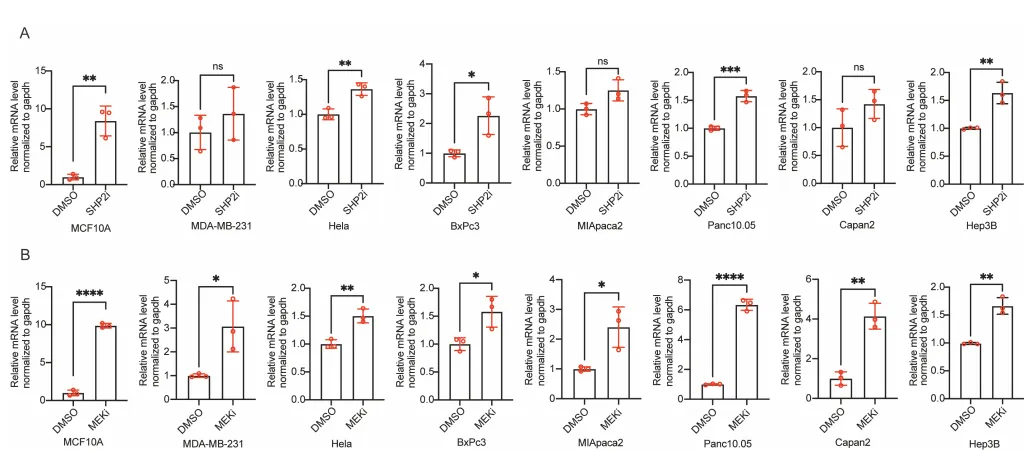
PS:亮点(直击工业界耐药痛点):只要 RAS-ERK 增殖信号被靶向药斩断,癌细胞就会疯狂吐出 CD133+ 囊泡。开启 Intercellsome 绝对不是特例,而是实体瘤对抗靶向药的高度保守的底层逻辑。
二、 转录组学的决定性证据,彻底与外泌体划清界限
如果 CD133 囊泡只是普通的外泌体(EVs),那这套耐药机制的创新性就荡然无存了。
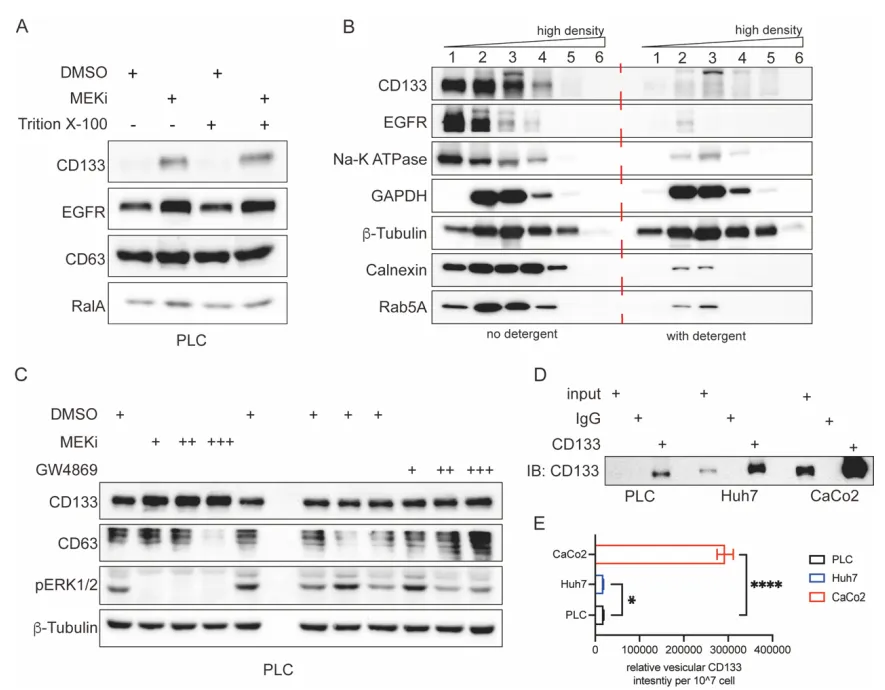
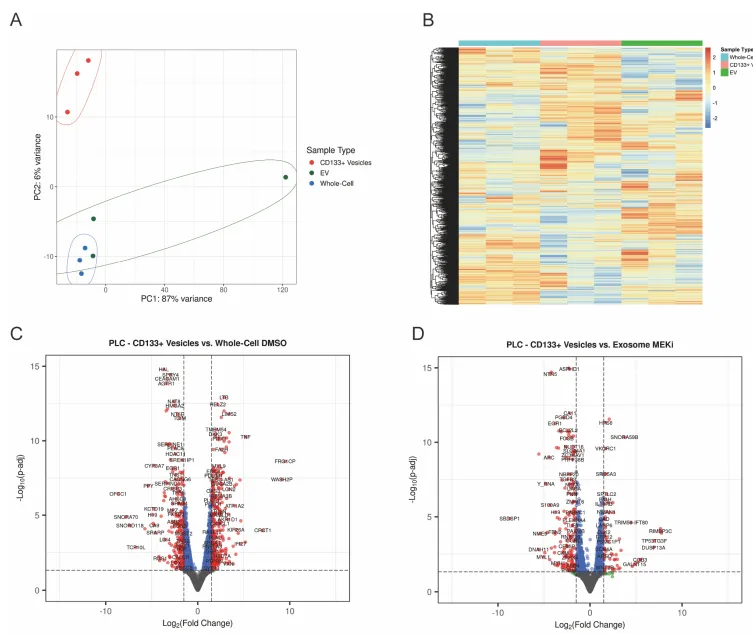
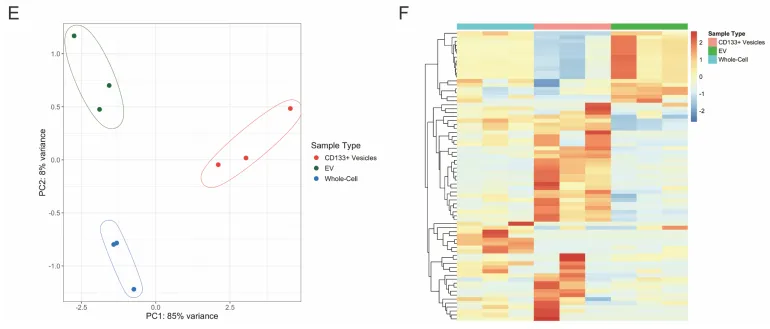
物理阻断。团队使用 GW4869(经典外泌体分泌抑制剂)处理细胞,普通外泌体标志物锐减,但 CD133 囊泡生成丝毫不受影响。相反,加入去污剂(Triton X-100)则会彻底破坏外泌体的脂质双分子层结构(Supp Fig 5)。
组学验证。对纯化出的 CD133+ 囊泡、普通 EVs 和全细胞进行 RNA-seq。主成分分析(PCA)和火山图极其清晰地显示:CD133+ 囊泡携带的 RNA 图谱与外泌体截然不同(Supp Fig 6)。它们绝不是往细胞外随机倾倒的垃圾袋,而是精准运送 mRNA 的走私盲盒。
PS:亮点(物理与组学的双重金标准铁证):利用去污剂破坏外泌体的脂质双层结构,结合 RNA-seq 揭示 RNA 货物图谱的巨大差异,以无可辩驳的双重铁证,彻底将 CD133+ 囊泡与传统外泌体划清界限。
三、 揪出幕后黑手:Hippo-YAP 通路是操纵暗网的总司令
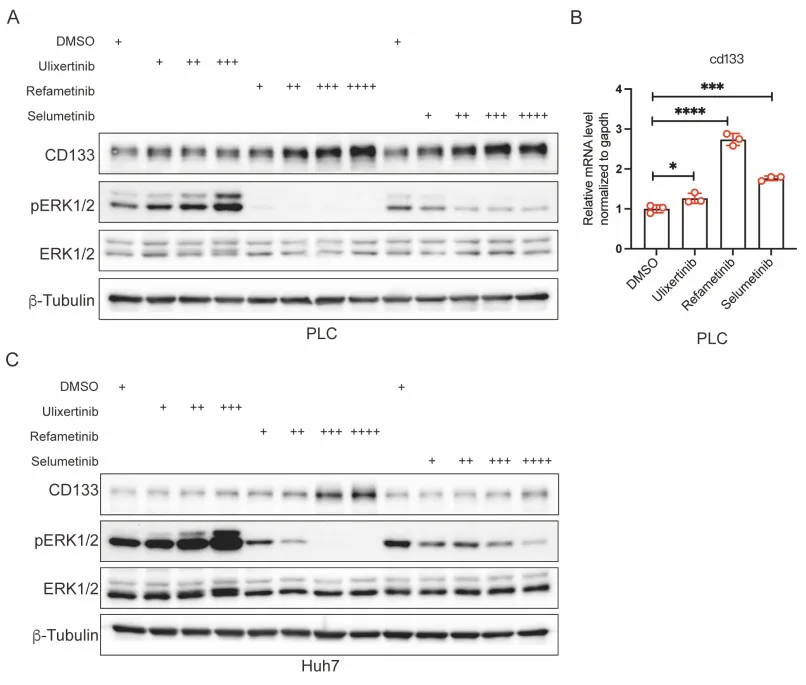
为什么 MEK 抑制剂会导致 CD133 飙升?团队顺藤摸瓜,找到了大名鼎鼎的耐药枢纽,Hippo-YAP 通路。
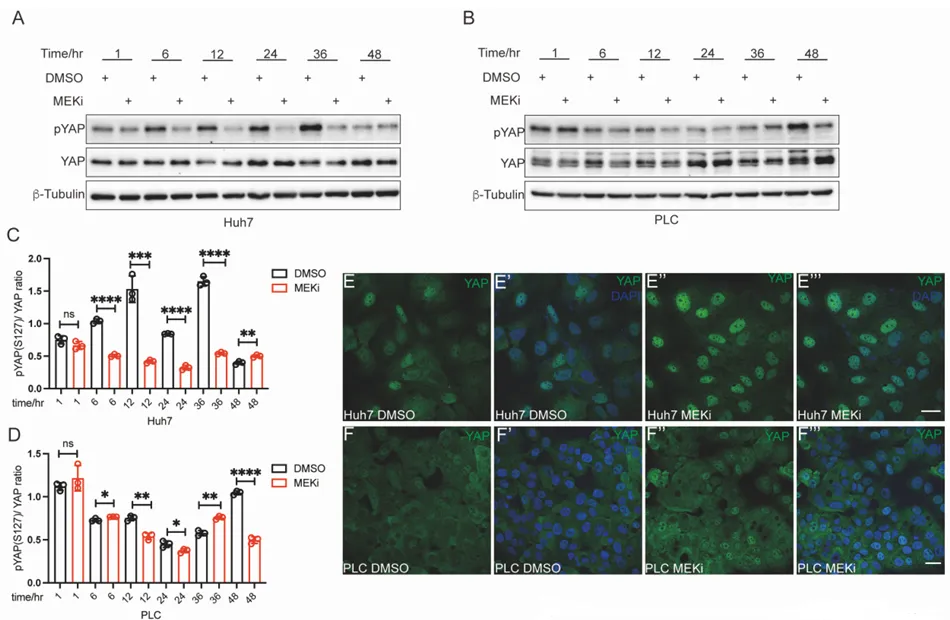
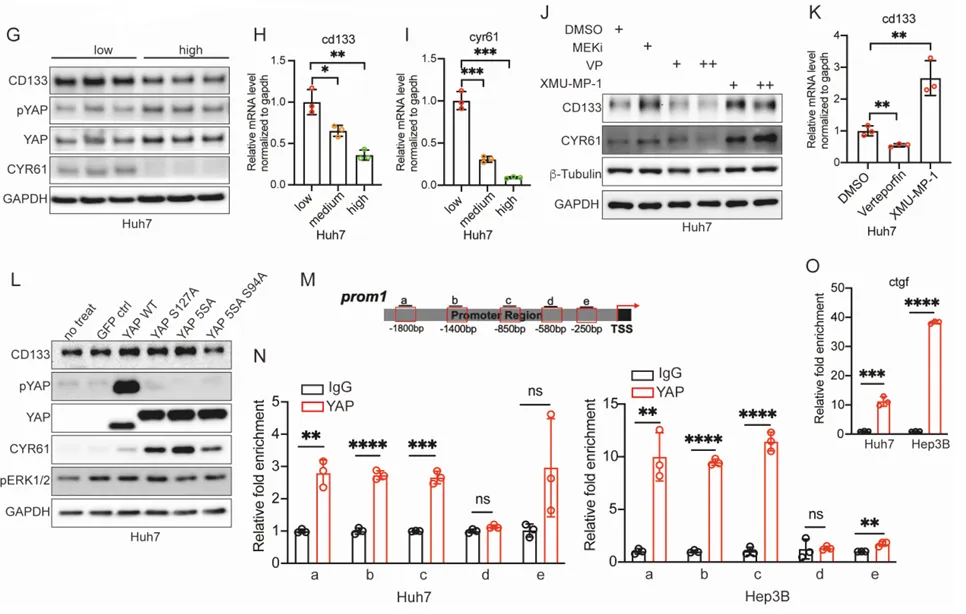
PS:亮点(ChIP-qPCR 的极致机制溯源):没有停留在蛋白表达的表面相关性上,而是通过染色质免疫沉淀技术,将 YAP 活化与 CD133 转录启动直接绑定,完美回答了耐药机制中谁在发号施令的核心问题。

🔹 什么是 Hippo-YAP 通路?这是一套决定细胞生死的“刹车与油门”系统。Hippo 激酶级联是“刹车”,限制细胞过度增殖;而 YAP 蛋白是“油门”,负责驱动细胞生长。
🔹 运作机制。正常状态下,刹车踩死,YAP 被磷酸化,在细胞质内被降解。但在绝境或癌变状态下,刹车失灵,未磷酸化的 YAP 冲进细胞核,唤醒耐药基因。
🎯 工业界痛点。当临床靶向药(如 EGFRi、MEKi)死死堵住主流增殖通道时,肿瘤细胞极易激活 YAP 进行旁路代偿。YAP 堪称多药耐药(MDR)的终极枢纽,这也是开发 YAP 抑制剂成为当下跨国药企前沿热点的原因。

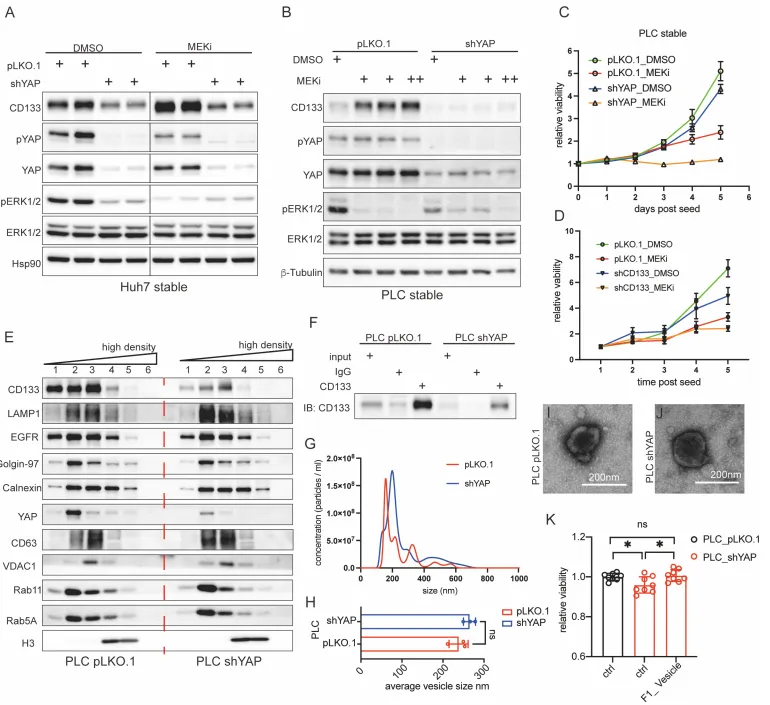
体外实验。团队利用 shRNA 敲低 YAP,癌细胞瞬间丧失抵抗力,对 MEKi 的敏感性大幅增加(增殖曲线彻底趴下)。但最绝妙的 Rescue 实验在于。如果在敲低 YAP 的濒死细胞中,人为地加入纯化好的 CD133+ 囊泡,细胞竟然奇迹般地复活了!这完美证明了 CD133 囊泡是 YAP 介导耐药的绝对核心执行者(Fig. 4)。
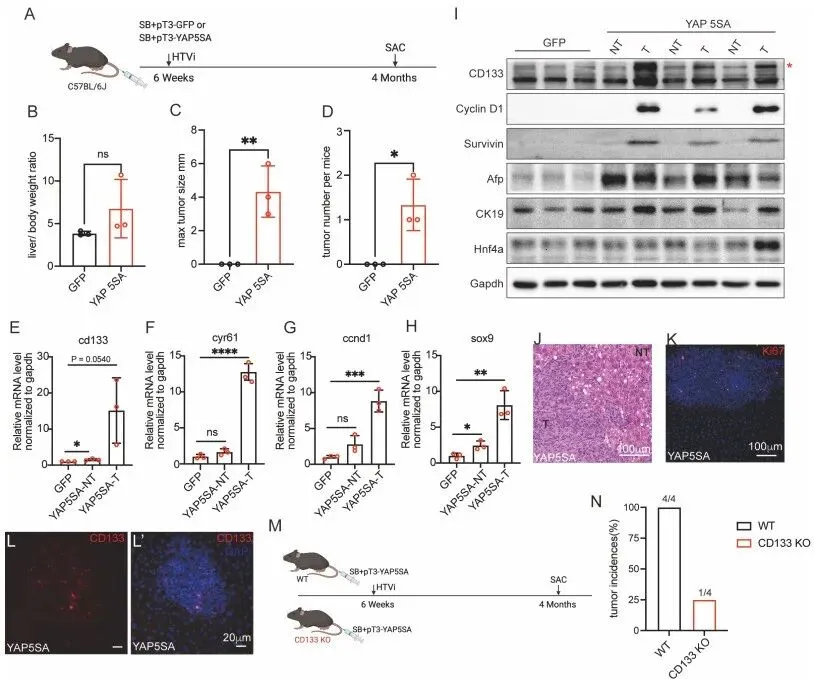
体内实验。团队利用尾静脉高压注射,在小鼠肝脏内转入持续激活的 YAP(YAP-5SA),成功诱导出巨大的肝癌。但震撼的是:当他们在 CD133 基因敲除(KO)的小鼠身上重复同样的实验时,肿瘤发生率暴跌,几乎无法成瘤!(Fig. 5)。在活体层面彻底盖棺定论。没有 CD133 搭建的走私网络,YAP 就算再疯狂,也无法独自支撑肿瘤的恶性扩张。
PS:亮点(Rescue 实验与活体成瘤的转化铁证):体外人工补充囊泡能让癌细胞起死回生,而体内敲除 CD133 直接让耐药肿瘤飞灰湮灭。这为未来开发靶向 YAP-CD133 轴的 Combo 疗法提供了极具爆发力的临床转化证据。
💡 结语,Intercellsome的转化价值
👆 探索更多转化医学底层逻辑,欢迎关注。我们继续前进!
如果你对感兴趣,推荐继续阅读:
拒绝生信内卷!牛津团队仅凭4个样本砸百万做空转,活体亚细胞空间组学重塑研发范式
从“强拆”到“重编程”:张泽民院士团队顶刊揭秘,寻找控制细胞命运的终极开关
1. Identification of CD133 intercellsomes in intercellular communication to offset intracellular signal deficit. eLife. 2023
2. CD133+ Vesicles Mediate Resistance to RAS-ERK Inhibition Regulated by YAP Activation in Liver Cancer Cells. bioRxiv. 2025
注:本文图文解读基于公开学术文献,文中图表均引自原论文,仅供学术与行业交流。

