药物发现是一个耗时且成本高昂的过程。传统的高通量筛选以及基于对接的虚拟筛选由于成功率低、可扩展性有限而受到制约。近年来,生成式建模的进展使得从头配体设计突破了枚举式筛选的限制。然而,这些模型往往存在泛化能力不足、可解释性有限的问题,并且过度强调结合亲和力而忽视关键的药理学性质,从而限制了其转化应用价值。
2025年12月18日,深圳大学朱泽轩与浙江大学侯廷军研究团队在arXiv上发表题为“Toward Closed-loop Molecular Discovery via Language Model, Property Alignment and Strategic Search”的研究论文。
研究提出一种分子生成框架Trio,将基于片段的分子语言建模、强化学习以及蒙特卡洛树搜索相结合,实现高效且可解释的闭环靶向分子设计。实验结果表明,Trio能够稳定地产生化学上有效且药理性质得到增强的配体,在结合亲和力、类药性和合成可及性方面均优于当前最先进的方法,同时将分子多样性提升至原来的四倍以上。通过融合泛化性、合理性与可解释性,Trio建立了一种闭环生成范式,重新定义了化学空间的探索方式,为下一代AI驱动的药物发现奠定了变革性的基础。
Trio代码仓库:
https://github.com/SZU-ADDG/Trio
药物发现是一项极其复杂、成本高昂且耗时漫长的系统性工程,通常需要持续十年以上的投入以及巨额资金,才能将单一治疗候选物转化为获得临床批准的药物。近年来,生成式建模使得在特定任务优化约束下设计新型先导化合物成为可能。
近期的研究引入了自回归生成模型,尝试直接从蛋白质三维结构上下文中设计配体。代表性方法包括Pocket2Mol、ResGen以及FragGen。尽管这些模型能够在分子生成过程中条件化蛋白特征,但其严格的序列化生成方式偏离了物理现实,误差在生成过程中不断累积,往往导致化学上不合理的结构。为克服上述问题,扩散模型和基于流的模型应运而生,它们通过同时生成所有原子,从而在生成过程中捕获全局相互作用。典型方法包括DiffBP、DiffSBDD以及EquiFM。总体而言,这些面向靶标的条件生成模型在生成高亲和力配体方面展现出良好潜力。然而,实验解析的蛋白–配体复合物数量有限,仍严重制约着模型训练,从而限制了其在实际药物发现应用中的泛化能力与鲁棒性。为克服蛋白条件生成模型在泛化能力方面的不足,研究人员日益从语言模型中汲取灵感,尤其是在GPT等模型于多个领域取得成功之后。代表性工作包括BindGPT、3DSMILES-GPT以及TamGen。尽管这些方法在一定程度上提升了模型的泛化能力,现有的分子语言模型在精确靶向蛋白结合口袋方面仍显不足。此外,辅助优化流程往往过度强调结合亲和力,而忽视类药性和可合成性等关键因素,从而限制了其在药物发现中的实际转化价值。
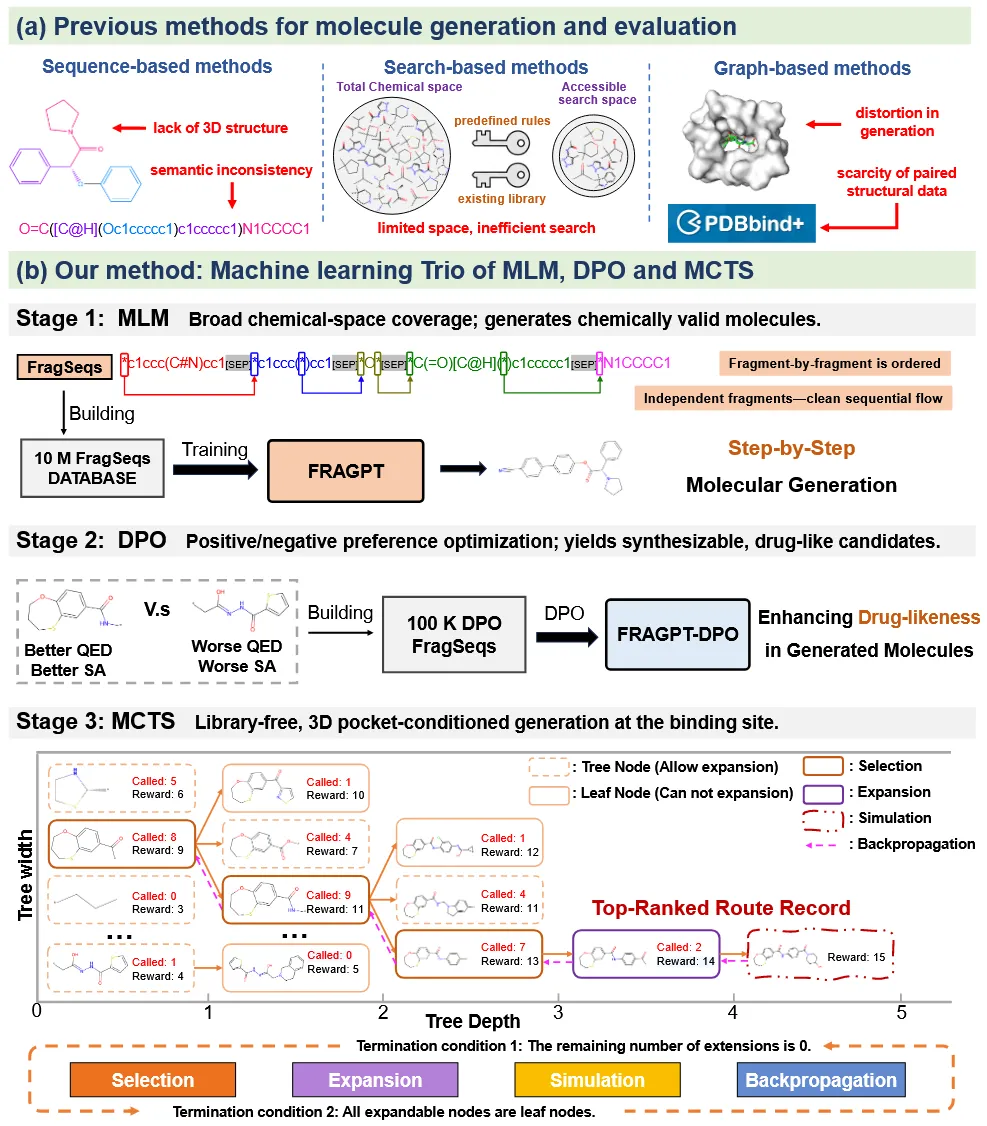
总而言之,尽管近年来的分子生成模型为探索化学空间和设计新型化合物提供了强有力的手段,但它们往往将分子设计简化为过度原子化或过度符号化的表示形式(图1a)。这类方法通常通过基于物理的优化来优先考虑与结合位点残基的局部相互作用,却忽视了分子功能在语义层面的整体一致性,从而削弱了分子亲和力在化学上的合理性。此外,现有模型可解释性不足仍是一个根本性障碍。其黑箱特性掩盖了分子优化的路径,使化学家难以对设计结果进行理性解释或建立信任,进而限制了其在药物发现中的广泛应用。
Trio整体生成流程可划分为三个阶段。阶段1:采用自监督学习训练分子语言模型(MLM),用于执行下一片段预测任务;阶段2:利用强化学习对MLM进行微调,以实现定制化的分子性质对齐;阶段3:结合蒙特卡洛树搜索(MCTS)与已对齐的MLM,在三维蛋白结合口袋中逐步生成分子结构。
Trio的监督式MLM采用类GPT的架构,命名为FRAGPT,以自回归方式预测分子片段。具体而言,用于训练的原始分子SMILES字符串需先转换为基于片段的SMILES标记序列。随后,监督式MLM通过因果注意力机制,基于上下文语义环境逐步生成分子片段,如图1b所示。Trio采用直接偏好优化(DPO)对监督式MLM进行微调,通过将模型的条件分布与反映目标分子属性的外部偏好信号进行显式对齐。经对齐后的MLM能够同时生成满足多项靶向性质要求的可成药分子。此外,Trio在复杂的靶标感知分子设计任务中,将对齐后的MLM与MCTS算法相结合。这种混合式方法充分利用了MCTS在探索与利用之间进行平衡的优势,从而促进具有更高结合亲和力且更加多样化的分子生成。该范式还具备较高的灵活性,可通过调整奖励函数直接改变搜索目标,从而避免重复微调模型所带来的计算开销。更重要的是,相较于单纯的微调方法,这种逐片段的搜索过程在可解释性方面具有显著优势:分子片段的优化轨迹能够直观反映策略决策过程,而基于神经网络权重的微调方法其可解释性往往受限于黑箱特性。
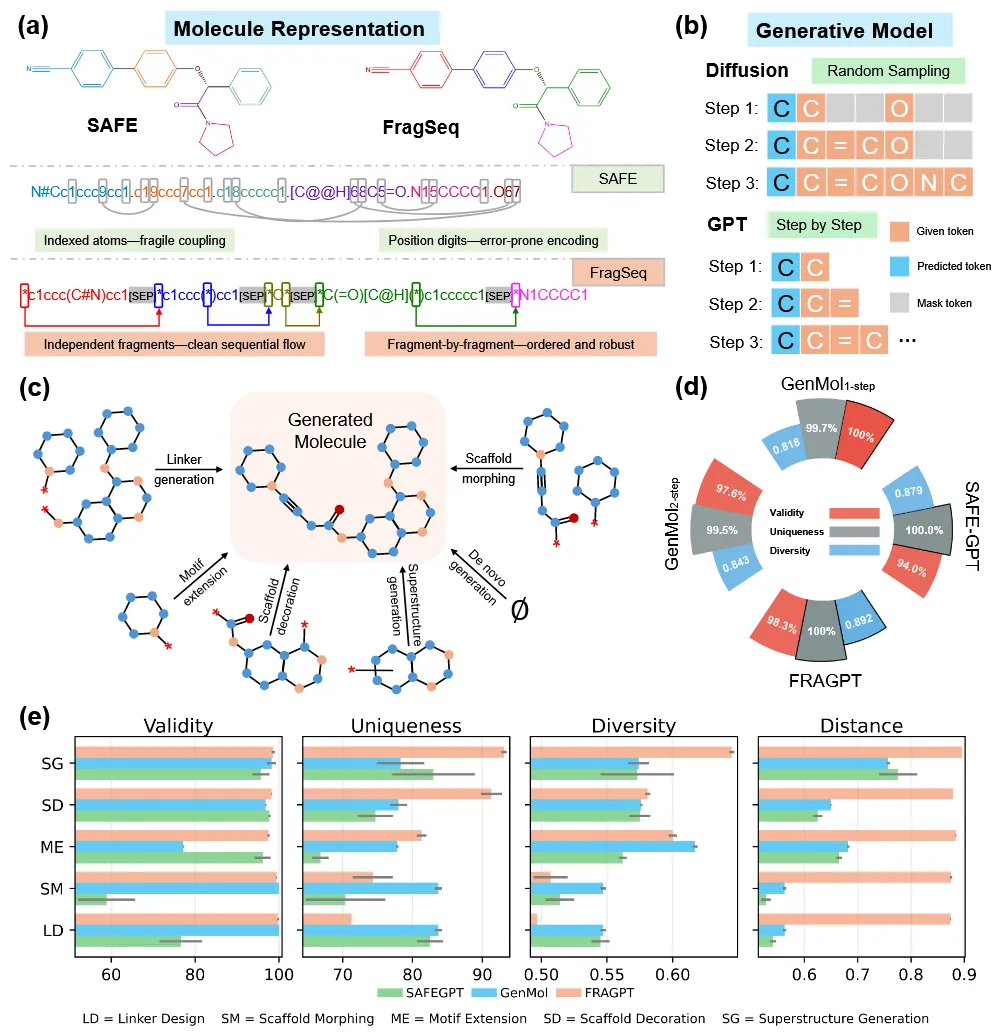
在从头分子生成任务中,仅使用SAFE数据集1%数据训练的FRAGPT,能够达到甚至超越在完整数据集上训练的基线模型性能,充分体现了其卓越的数据效率(图2d)。SAFEGPT相对较低的有效性主要源于其依赖位置相关的数值标记进行片段连接;随着片段数量的增加,这些数字会干扰规范的环闭合标记,从而显著提升语法歧义并导致可扩展性不足。FRAGPT通过引入结构化的片段语法,将连接位点语义与环索引解耦,有效规避了这一失效模式,从而同时实现了更高的有效性和更丰富的结构多样性。尽管基于扩散模型的GenMol在有效性方面表现较好,但其相对保守的去噪调度抑制了化学空间探索,导致生成分子的多样性不足。
图2 FRAGPT在从头生成与片段约束分子生成中的表示方式、模型架构、任务设置与性能表现。
在评估片段约束分子设计性能时,作者围绕五类关键任务对FRAGPT进行了严格评估,包括骨架修饰、骨架变换、连接子生成、基序扩展以及超结构生成。图2e展示了在每个任务中生成100个样本的片段约束生成结果。结果表明,FRAGPT在所有任务中均实现了接近完美的有效性,并在结构距离指标上全面领先。即便在结构约束最为严格的连接子设计和骨架变换任务中,FRAGPT仍展现出显著的生成多样性,其分子间距离明显高于所有对比方法,说明生成候选分子覆盖了更加遥远且新颖的化学空间区域。在约束程度较低的任务(如基序扩展、骨架修饰与超结构生成)中,FRAGPT同样稳定实现了高唯一性、大范围探索能力以及良好的化学保真度,体现了模型在灵活性与精确性之间的优异平衡。
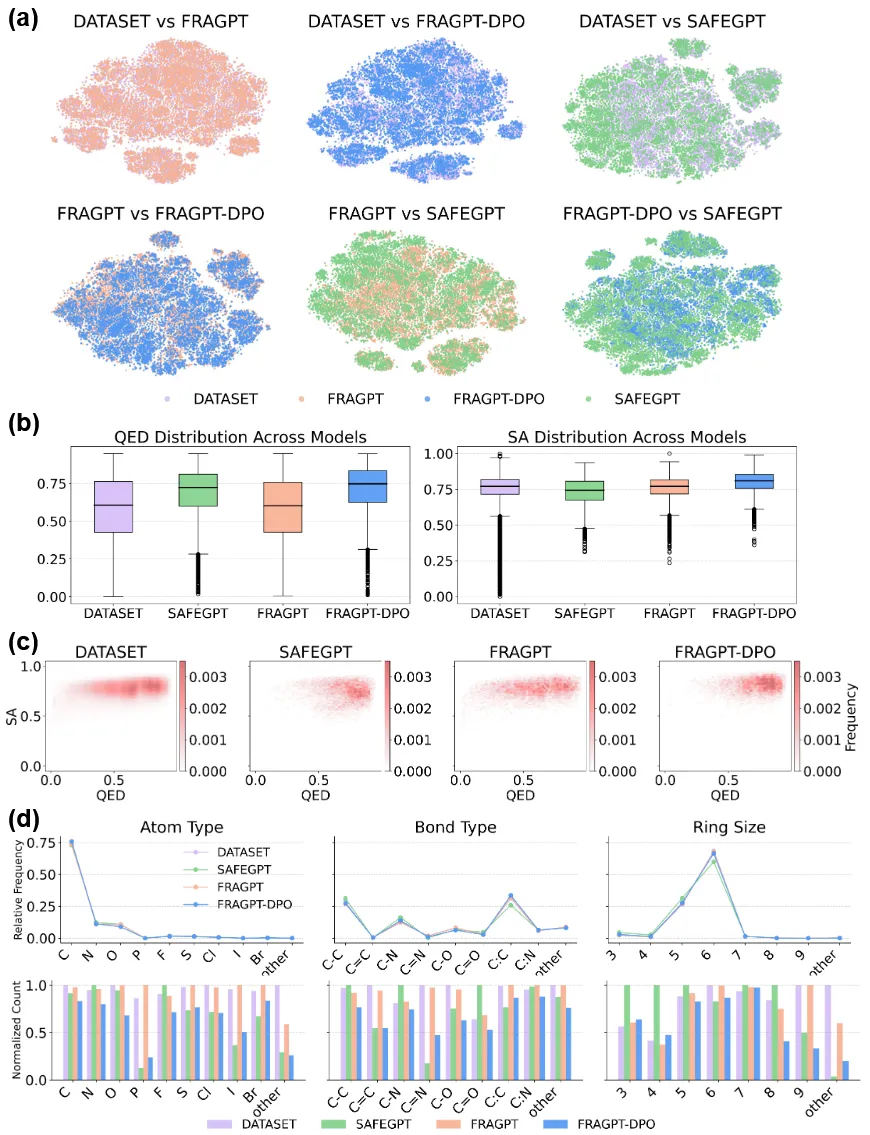
研究团队进一步采用DPO算法,将FRAGPT模型与类药性评分进行对齐,为后续的靶标特异性分子生成任务做准备。如图3a所示,原始FRAGPT几乎完全覆盖了训练数据的分布流形。FRAGPT-DPO倾向于压缩既有分布并将样本密度向内部聚集,而SAFEGPT则产生了多个在FRAGPT-DPO分布景观中不存在的新高密度簇。
图3 基线数据与不同生成模型所生成化学空间表征对比
从图3b和图3c可以看出,原始FRAGPT在QED-SA联合分布上与训练数据高度一致。与此同时,SAFE在QED指标上相较原始FRAGPT有所提升,但其SA分布更为分散,表明其在优化过程中更偏向类药性而牺牲了一定的合成可及性。经DPO对齐后,FRAGPT-DPO的QED分布出现明显上移,SA亦呈现出适度提升,同时SA的方差显著收缩。如图3d所示,上方面板表明三种生成模型在原子类型、键类型和环尺寸分布方面均能较好地复现训练集的统计特征。下方面板进一步显示,原始FRAGPT在上述三类描述符上均保持了与数据集相近的频率分布。尽管这一特性扩展了结构多样性,但也导致生成分子的SA和QED得分下降。FRAGPT-DPO在生成过程中主动舍弃了化学上不利的结构基元,从而在类药性和合成可及性方面相较于原始数据集实现了显著提升。
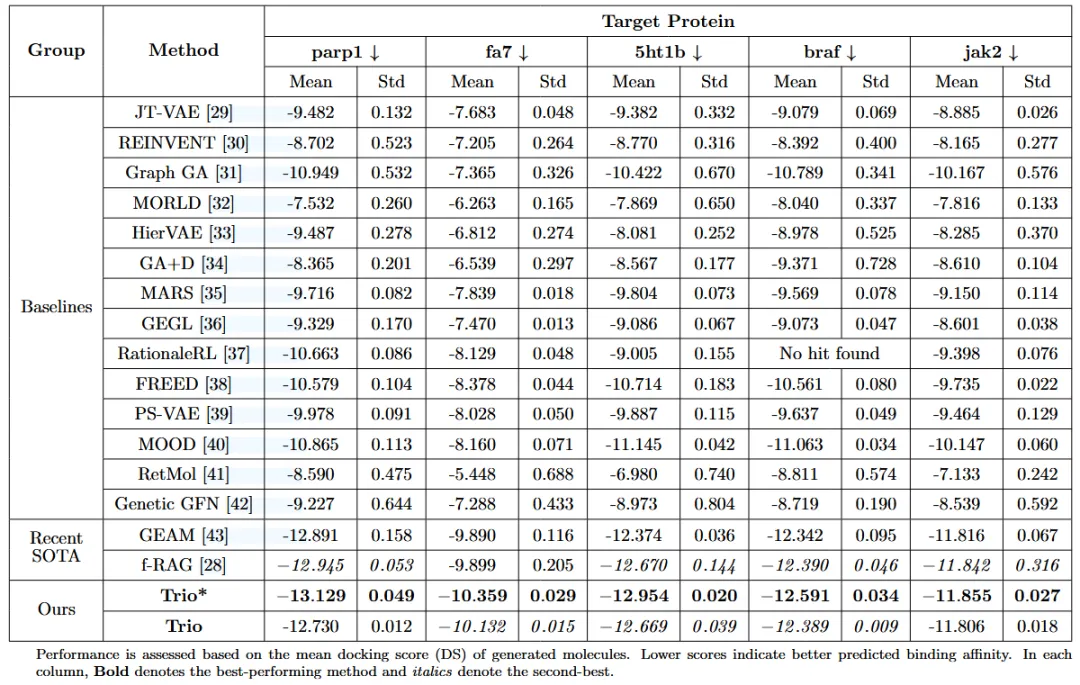
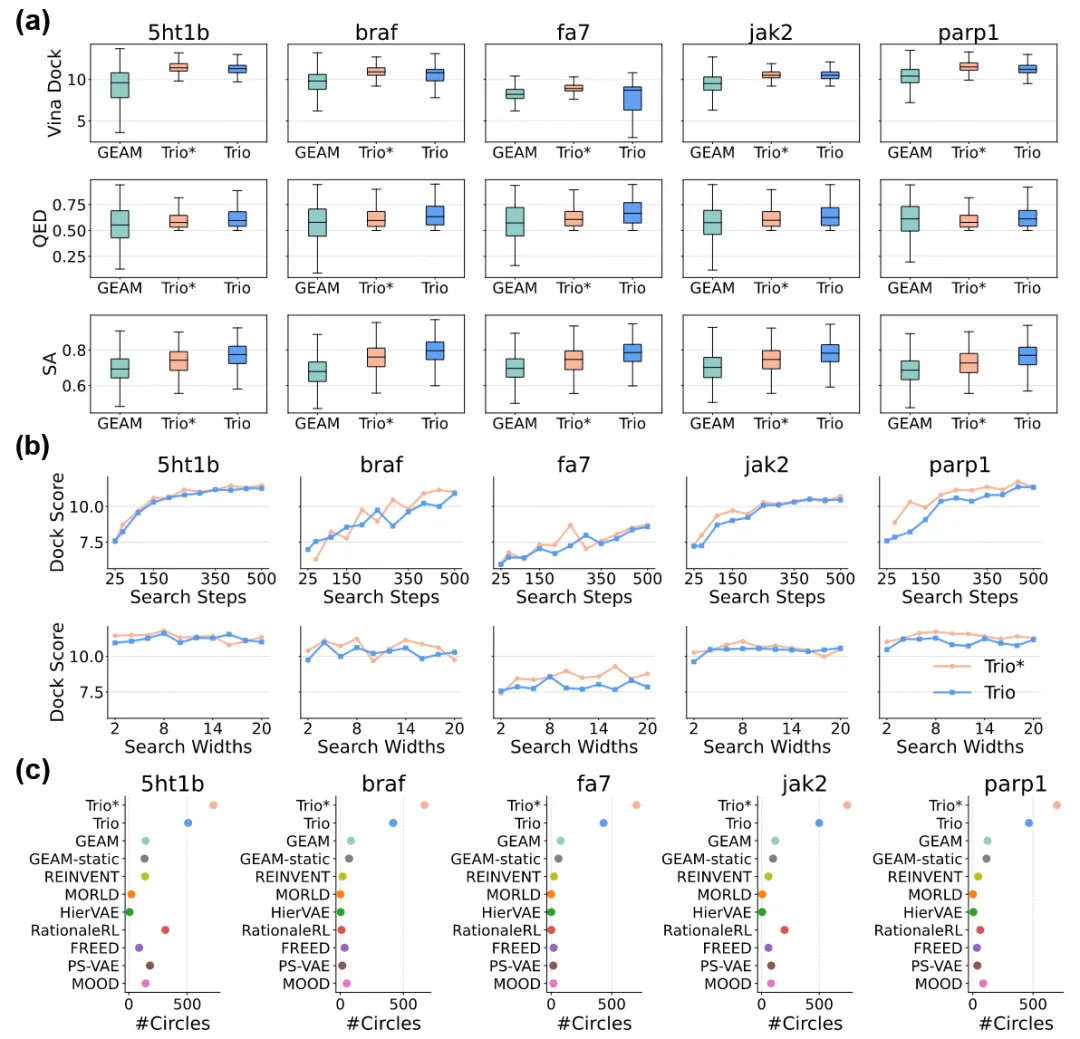
针对每个蛋白靶标,生成了3000个候选分子,并与最先进的基线生成模型进行了对比评测。如表1所示,基础版Trio*模型(未引入DPO约束)在五个靶标上均取得了最佳的结合亲和力,整体性能显著优于所有对比方法。
完整的Trio框架将FRAGPT-DPO与MCTS深度融合,构建了一种面向类药分子搜索的整体性解决方案。为避免由高度相似分子簇导致的性能虚高,作者对模型生成的分子进行去冗余处理。即便在去除结构冗余之后,Trio*与Trio仍分别保留了超过70%的候选分子,凸显了二者在生成广度方面的显著优势。如图4a所示,完整Trio模型在偏好对齐机制的驱动下,在QED和SA指标上取得了更优且更为集中的分布,从而为实际药物发现提供了最优的综合平衡。如图4b所示,随着模拟次数的增加,对接评分整体呈现改善趋势;相比之下,单纯扩大树宽度虽能增强探索性,但并未带来具有统计显著性的对接性能提升。
如图4c所示,Trio*在五个蛋白靶标上均实现了#Circles指标的显著多倍提升,反映了其几乎不受约束的探索能力。完整Trio模型由于引入偏好对齐约束,其#Circles相较Trio* 略有下降,但仍明显优于以往方法。这一显著提升表明,作者提出的方法能够有效突破基于规则搜索和静态片段库的固有限制,实现更加多样且新颖的分子生成。尤为重要的是,这一优势在所有靶标上均保持一致,与受体类型或结构复杂性无关,说明MLM与树搜索的结合在不同生物学背景下具有稳健的泛化能力。这种一致性规避了纯数据驱动或规则受限方法中常见的靶标迁移性问题,充分展示了Trio在多样化化学空间中进行高效导航的独特适应性。
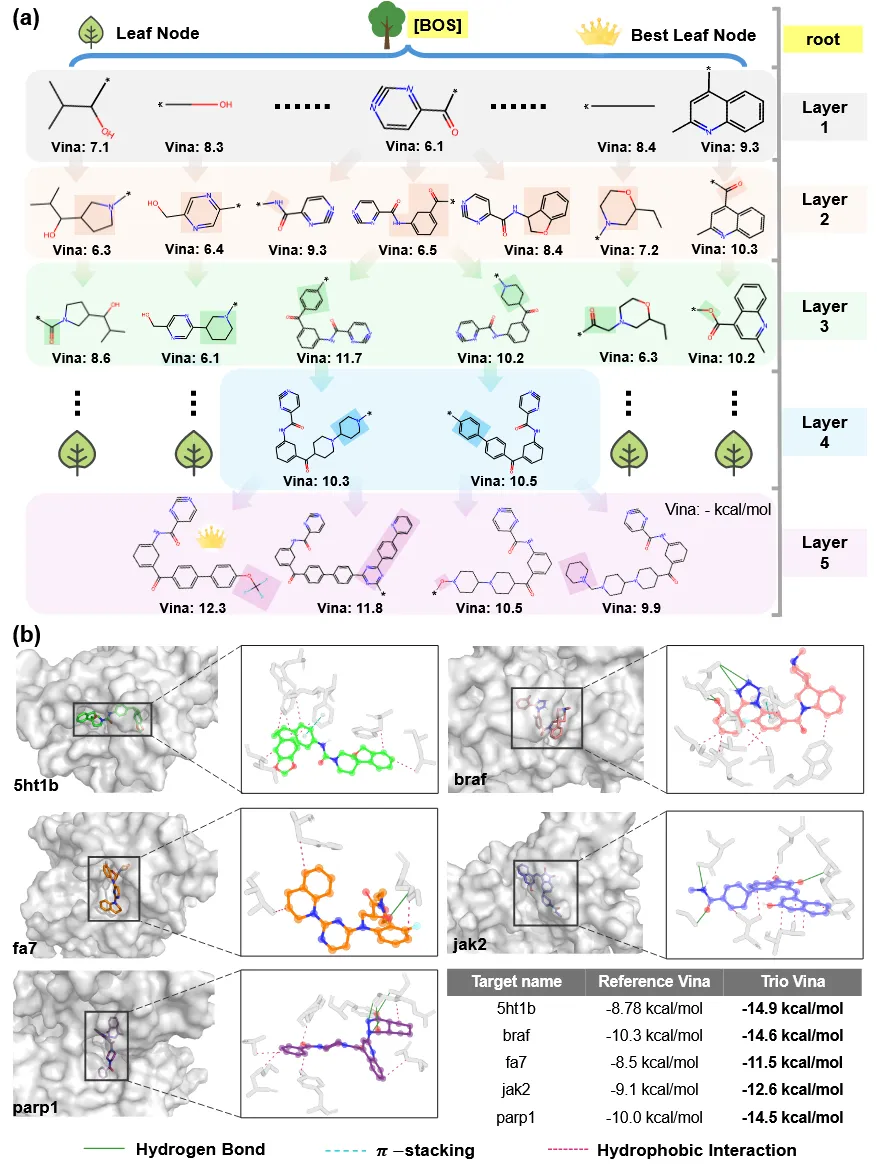
图5 Trio框架逐步生成机制及生成配体与靶蛋白结合口袋间分子相互作用的示意图。
如图5a所示,Trio的协同式架构被刻画为一种由MCTS引导的层级化搜索过程,其将FRAGPT中编码的大规模语义知识与MCTS久经验证的搜索效率无缝融合。这种可解释的设计范式为药物化学家提供了可操作的洞见,支持一种更加理性、以人为中心的工作流程,有效连接生成模型与专家驱动的药物发现实践。图5b中的对比表显示,Trio生成配体在多个靶标口袋中的Vina评分显著优于参考化合物,平均提升幅度达46.0%。这一系统性的相互作用剖析充分佐证了该模型在生成化学上有效、可合成且具备更高特异性与预测亲和力的配体方面的能力。
Trio体现了AI驱动药物发现的一次范式转变:它重塑了化学空间的探索方式,使系统化、多目标的搜索过程既具备可解释性,又能够在实践中落地。未来的拓展方向包括引入逆合成推理、更复杂的ADMET感知奖励函数以及更丰富的片段词表,从而进一步提升该框架应对以往难以攻克的生物靶标的能力。最终,Trio为自主化、闭环式药物发现奠定了基础,勾勒出迈向新一代理性化、AI引导治疗药物研发的清晰路径。
参考链接:
https://doi.org/10.48550/arXiv.2512.09566
感兴趣的读者,可以添加小邦微信加入读者实名讨论微信群。添加时请主动注明姓名-企业-职位/岗位或姓名-学校-职务/研究方向。