作者
陈文(深圳大学)
骆静利(深圳大学)
引言
甲醇作为一种廉价液体燃料(约350美元/吨),具有高能量密度(17.28 MJ/L)和易于储存运输的特点,可通过现有汽油基础设施进行大规模配送。甲醇氧化反应在直接甲醇燃料电池(DMFC)和替代氧析出反应的节能氢气生产中具有重要应用前景。然而,当前甲醇氧化技术面临多重挑战:Pt基催化剂虽然活性较高,但存在CO中间体中毒导致活性位点快速失活、高起始电位(约450 mV)以及CO2排放等问题;Ni基催化剂虽然可生产高附加值甲酸盐(约1300美元/吨),但通常需要高过电位(>1.3 V)。此外,传统催化剂难以同时实现高活性、高选择性和长期稳定性,特别是在工业级电流密度下的应用受限。因此,开发兼具低过电位、高甲酸盐选择性、抗CO中毒能力和优异稳定性的高效甲醇氧化催化剂,对于推动DMFC实用化和节能氢气生产至关重要。
核心发现
本研究通过一锅法热分解策略成功合成了单分散的金属间化合物PtBi核/拉伸应变PtNiBi壳(I-PtBi@TS-PtNiBi)纳米片催化剂。该催化剂在碱性电解质中展现出优异的甲醇氧化性能,质量活性达到33.2 A mg⁻¹ Pt,分别是PtBi(5.1 A mg⁻¹ Pt)、PtNi(3.1 A mg⁻¹ Pt)和Pt/C催化剂(4.2 A mg⁻¹ Pt)的6.5倍、10.7倍和7.9倍。在0.6至1.2 V的宽电位窗口内,该催化剂实现了高甲酸盐法拉第效率,避免了CO2排放。基于该催化剂构建的DMFC器件达到175.0 mW cm⁻²的峰值功率密度,甲醇辅助电解池在1.0 A cm⁻²电流密度下仅需0.79 V的电池电压,显著低于传统水电解。催化剂展现出优异的抗CO中毒能力和长期稳定性,在20,000秒测试后仍保持81.8%的初始活性,通过简单刷新电解质可实现140,000秒的稳定运行。DFT计算揭示了拉伸应变和NiBiOOH修饰协同作用使催化剂具有更高的甲酸盐选择性,有效避免了CO中毒问题。
图文解读
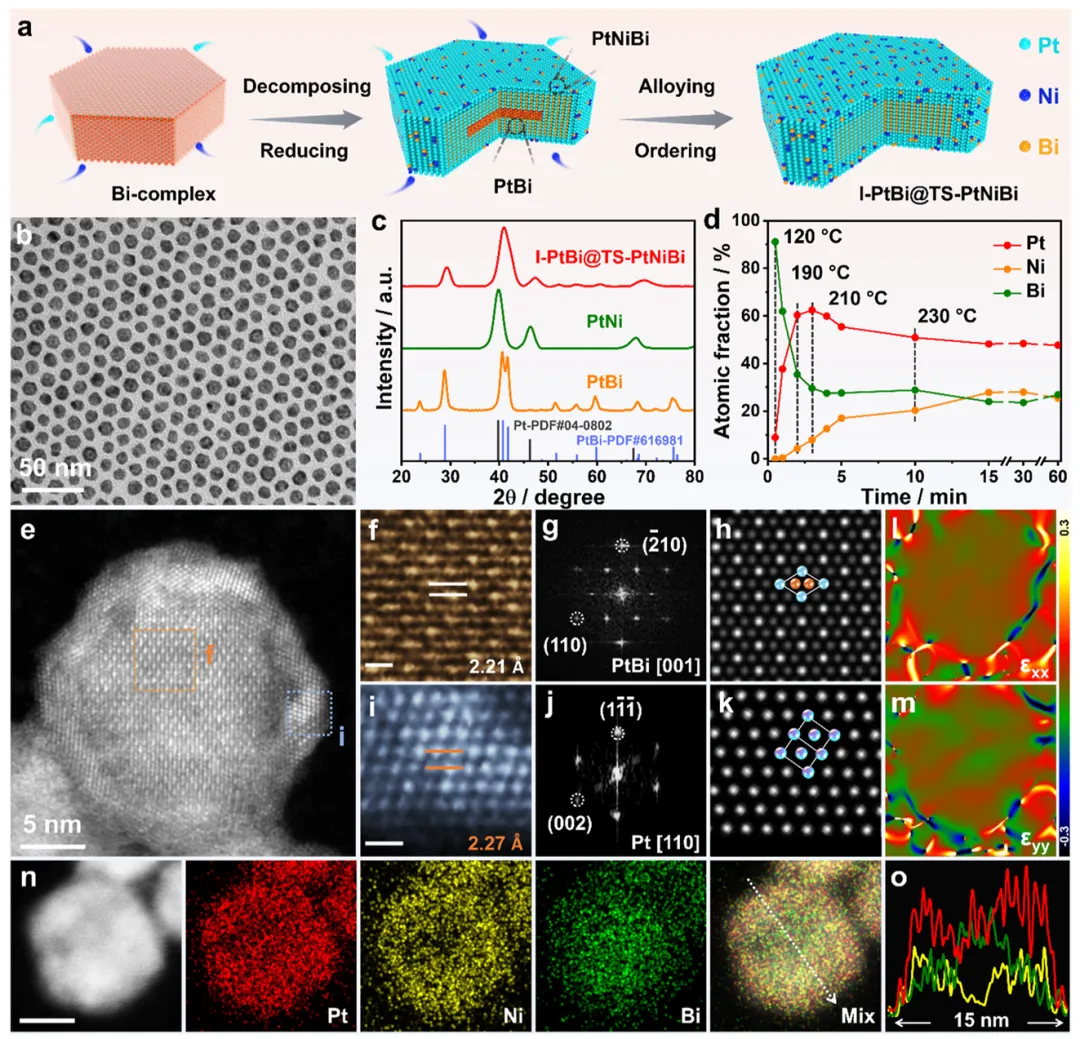
图1:I-PtBi@TS-PtNiBi核壳纳米片的合成与结构表征
图1展示了I-PtBi@TS-PtNiBi核壳纳米片的制备流程和结构特征。通过一锅法热分解策略,基于hcp-PtBi和fcc-PtNi的不同合金化行为,成功实现了PtBi金属间化合物核和PtNiBi三元合金壳层的同步形成。TEM和HAADF-STEM图像显示纳米片呈现单分散的核壳结构,平均粒径为11.3 nm,ICP-MS测定Pt/Ni/Bi原子比为48.6/28.2/23.2。XRD图谱证实了复合晶体相结构,融合了hcp-PtBi金属间化合物和fcc-Pt的衍射特征,fcc Pt基衍射峰向高2θ值偏移,源于多金属合金化或应变效应。时间依赖的结构演化研究表明,反应0.5分钟时形成Bi复合物模板上负载Pt团簇的无定形结构;1分钟后fcc Pt(111)衍射峰开始出现;4分钟后Bi复合物衍射峰消失,形成稳定的核壳结构。原子分辨率HAADF-STEM图像揭示了核区hcp-PtBi的有序原子排列,晶格间距2.21 Å对应PtBi(210)面;壳层fcc-PtNiBi(111)面晶格间距为2.27 Å,表明存在4.6%的拉伸应变。几何相位分析显示核壳界面存在压缩应变,壳层内部为拉伸应变。EDX元素分布图证实Pt均匀分布、Ni集中于壳层、Bi主要位于核区,验证了I-PtBi@TS-PtNiBi核壳纳米结构的成功构建。
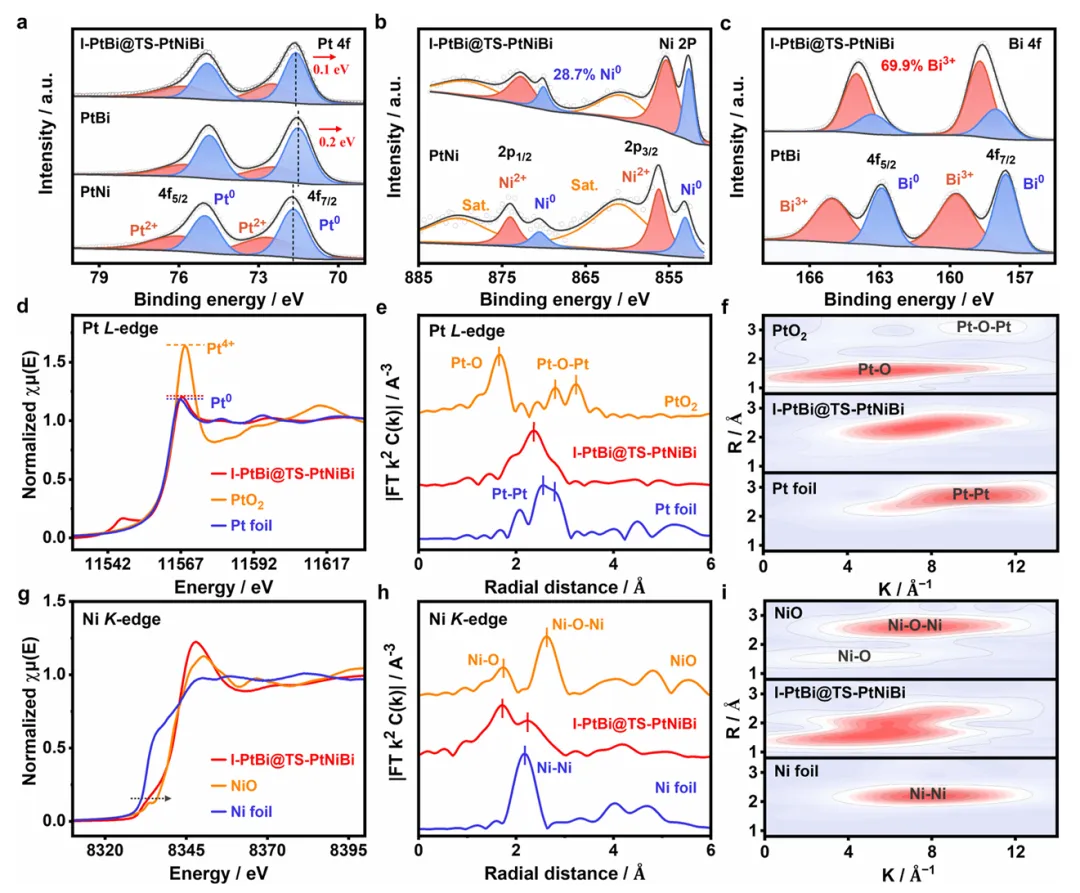
图2:I-PtBi@TS-PtNiBi的电子结构和化学状态分析
图2通过XPS和XAS技术深入分析了I-PtBi@TS-PtNiBi的电子结构和化学状态。XPS谱图显示Pt 4f、Ni 2p和Bi 4f均存在金属态和氧化态的共存,表明多金属Pt-Ni-Bi合金化特征,Ni和Bi的氧化态主要源于表面亲氧原子的暴露。I-PtBi@TS-PtNiBi中Pt0的结合能(71.6 eV)与PtBi(71.5 eV)和PtNi(71.7 eV)略有偏移,证实Pt、Ni、Bi原子间的三元电子相互作用。与PtBi金属间化合物(55.2%)相比,I-PtBi@TS-PtNiBi中Bi3+氧化态比例显著增加(69.9%),表明薄fcc-PtNiBi壳层中Bi3+为主导状态。XANES分析确认I-PtBi@TS-PtNiBi中Pt主要为Pt0态,Ni和Bi分别为Ni2+和Bi3+态。FT-EXAFS谱图显示Pt在I-PtBi@TS-PtNiBi中的最近邻原子环境与Pt箔和PtO2不同,主散射峰位于2.36 Å,低于Pt箔(2.55 Å),表明Pt-Ni合金配位导致的Pt-Pt键长缩短。WT-EXAFS轮廓图显示I-PtBi@TS-PtNiBi在7.5 Å⁻¹处出现强度最大值,对应Pt-Ni配位。Ni元素的FT-EXAFS散射峰位于1.75 Å和2.33 Å,分别对应Ni-O和Ni-Pt配位,WT-EXAFS强度最大值对应Ni-Pt和Ni-O路径。这些结果证实了I-PtBi@TS-PtNiBi中主导的Pt-Ni合金配位特征,为理解其优异催化性能提供了电子结构依据。
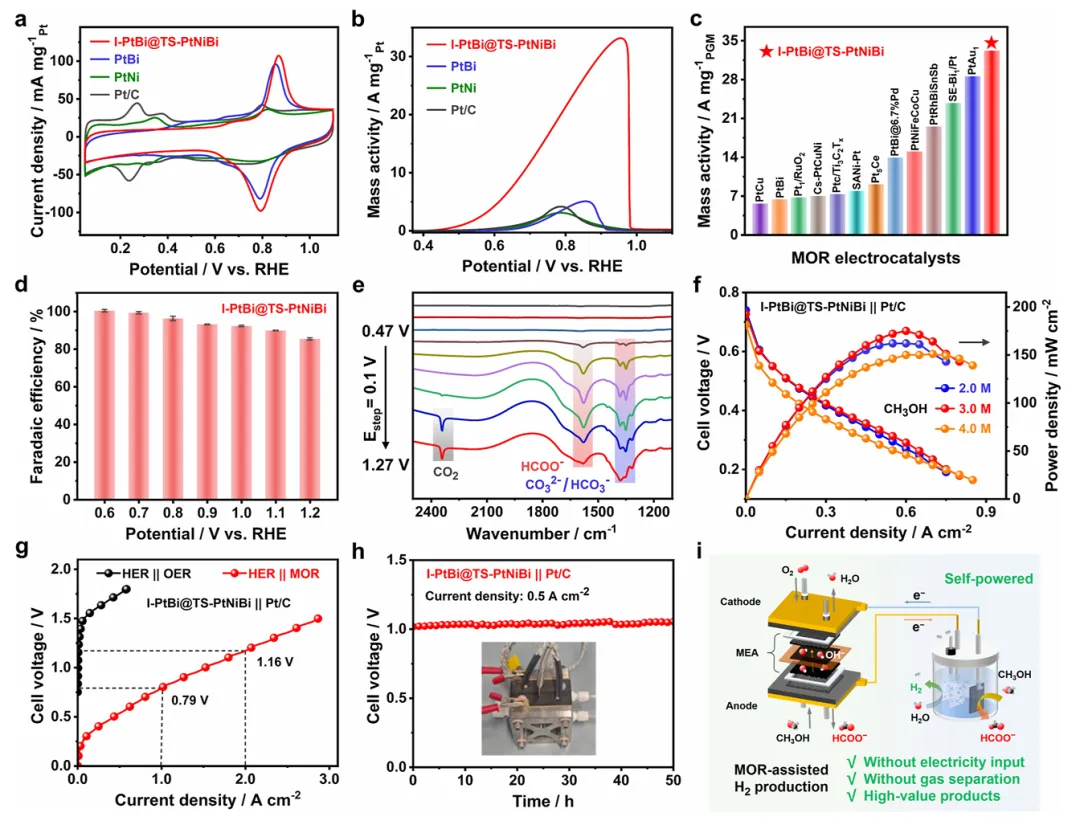
图3:I-PtBi@TS-PtNiBi的电催化性能与应用验证
图3系统评估了I-PtBi@TS-PtNiBi的甲醇氧化电催化性能及实际应用潜力。CV曲线显示,与Pt/C和PtNi催化剂相比,I-PtBi@TS-PtNiBi和PtBi催化剂的氢吸附/脱附峰(0.05-0.4 V vs RHE)明显抑制,源于表面Bi物种的暴露。正向甲醇氧化极化曲线显示,I-PtBi@TS-PtNiBi在碱性电解质中展现出33.2 A mg⁻¹ Pt的高质量活性,分别是PtBi(5.1 A mg⁻¹ Pt)、PtNi(3.1 A mg⁻¹ Pt)和Pt/C(4.2 A mg⁻¹ Pt)的6.5倍、10.7倍和7.9倍。该质量活性优于文献报道的多数碱性甲醇氧化催化剂。在0.6至1.2 V电位窗口内,I-PtBi@TS-PtNiBi实现了高甲酸盐法拉第效率,避免了CO2排放。原位FTIR谱图证实了甲酸盐的高选择性生成路径。基于I-PtBi@TS-PtNiBi构建的DMFC器件在不同甲醇浓度下均展现出优异性能,峰值功率密度达到175.0 mW cm⁻²。甲醇辅助阴离子交换膜水电解池在6.0 M KOH + 3.0 M CH3OH电解质中,仅需0.79 V电池电压即可达到1.0 A cm⁻²电流密度,显著低于传统水电解。在0.5 A cm⁻²电流密度下稳定运行,展现出优异的长期稳定性。该系统实现了DMFC和电解池双端甲酸盐生产,为自供电氢气生产提供了创新解决方案。
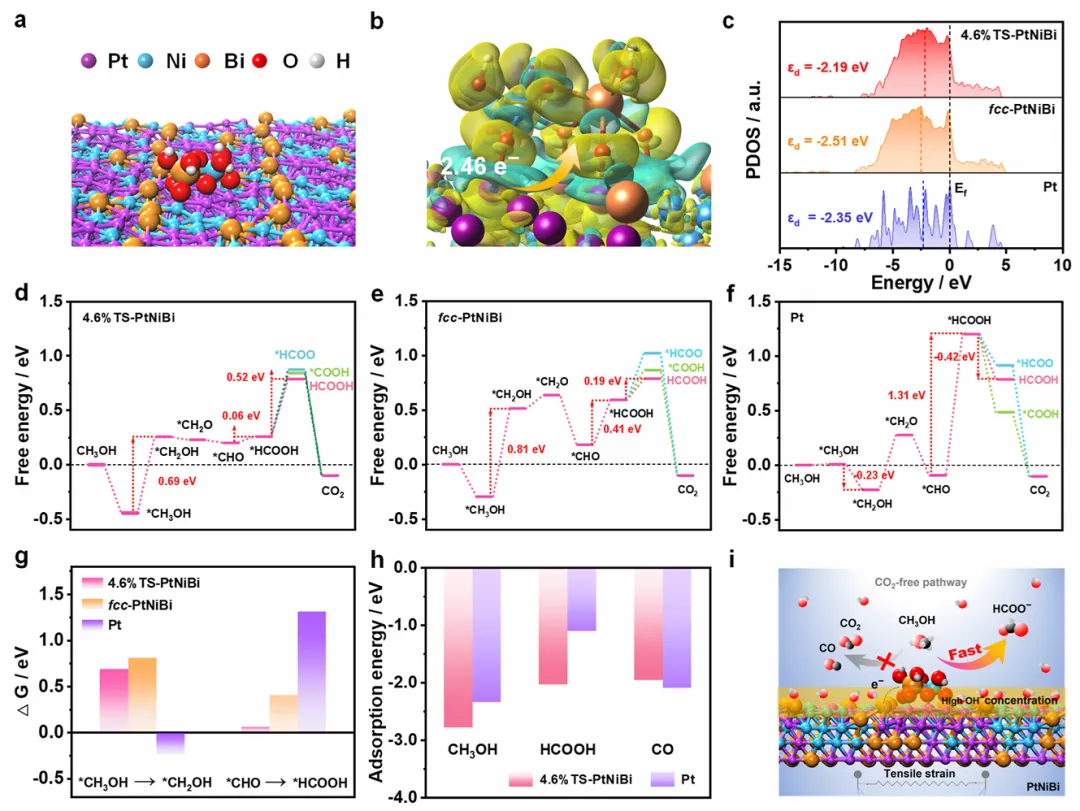
图4:DFT计算揭示拉伸应变增强甲醇氧化机理
图4通过DFT计算深入揭示了拉伸应变和NiBiOOH修饰对甲醇氧化性能的增强机理。计算模型包括4.6%拉伸应变PtNiBi(4.6%TS-PtNiBi)、无应变fcc-PtNiBi和Pt表面。d带中心分析显示,4.6%TS-PtNiBi具有更正的d带中心(εd = -2.19 eV),接近费米能级,相比fcc-PtNiBi(εd = -2.51 eV)和Pt(εd = -2.35 eV),该特征有利于更易断裂C-H或C-O键,降低氧化步骤的过电位,从而增强甲醇氧化活性。自由能图计算了甲醇氧化过程中各中间体的能量变化,包括*CH3OH、*CH2OH、*CH2O、*CHOH、*CHO、*HCOO、*COOH和*HCOOH等关键步骤。结果显示,*CH3OH转化为*CH2OH是4.6%TS-PtNiBi(ΔG = 0.69 eV)和fcc-PtNiBi(ΔG = 0.81 eV)的速率决定步骤,而*CHO转化为*HCOOH是Pt表面的速率决定步骤(ΔG = 1.31 eV),证实4.6%TS-PtNiBi具有增强的甲醇氧化活性。对于4.6%TS-PtNiBi和fcc-PtNiBi,*HCOOH脱附生成甲酸盐比转化为*COOH或*HCOOH更有利,表明其高甲酸盐选择性。相反,Pt表面*HCOOH转化为*COOH比脱附更有利,解释了Pt/C催化剂的高CO2选择性。吸附能分析显示,4.6%TS-PtNiBi对CH3OH和HCOOH的吸附比Pt更强,支持其改进的甲醇氧化活性和甲酸盐选择性。同时,4.6%TS-PtNiBi对CO的吸附不如Pt有利,表明更强的抗CO中毒能力。这些计算结果揭示了拉伸应变和NiBiOOH修饰协同作用使催化剂实现高甲酸盐选择性并消除CO中毒问题的机理。
总结
本研究成功实现了一锅法构建单分散hcp-PtBi核@fcc-PtNiBi壳纳米片,通过同步有序化-合金化机制形成具有4.6%拉伸应变的PtNiBi壳层。I-PtBi@TS-PtNiBi作为甲醇氧化催化剂展现出优异性能,质量活性达33.2 A mg⁻¹ Pt,实现高甲酸盐选择性,并具有强抗CO中毒能力和长期稳定性。原位FTIR、ATR-SEIRA和DFT计算验证了催化剂的高甲酸盐选择性和抗CO中毒机理。基于该催化剂开发的DMFC器件达到175.0 mW cm⁻²功率密度,甲醇辅助电解池在1.50 V下实现2.9 A cm⁻²电流密度。该自供电氢气生产系统为高效能量转换和液体燃料升级提供了创新解决方案,为原子级精准设计合成复杂核壳电催化剂用于高效能源转换提供了新见解。
原文链接
https://doi.org/10.1021/acsnano.6c05471


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
