钠离子混合电容器(SHCs)因传统负极材料中迟缓的钠离子动力学而受到严重制约。单原子位点虽然前景广阔,但其局部化的d轨道会引发过强的Na⁺结合,造成动力学瓶颈。
2026年02月05日,王振波、深圳大学隋旭磊团队在Angewandte Chemie International Edition期刊发表题为“FeCoN6 Sites Unlock Superior Sodium-Ion Storage Through Synergizing Capture-Release and Orbital-Mediated Charge Delocalization”的研究论文,团队成员Feng Wenliang、Xu Huifang为论文共同第一作者,王振波、隋旭磊为论文共同通讯作者。
第一作者:Feng Wenliang、Xu Huifang
通讯作者:王振波、隋旭磊
通讯单位:深圳大学
论文DOI:10.1002/anie.202525480
该研究设计了异核Fe-Co双原子位点(FeCoN₆),实现了快速且持久Na⁺存储的双重机制协同效应。理论与实验证据证实,强d-d轨道耦合诱导了轨道介导的电荷离域效应,该效应使d带中心下移,从而调节Na⁺的结合亲和力。同时,FeCoN₆位点固有的异核特性为Na⁺的捕获-释放路径提供了优化的阶梯式能量景观。这种电子轨道介导电荷离域调制与动力学捕获-释放模型之间的协同作用显著降低了扩散势垒。因此,Fe-Co双原子氮掺杂碳负极展现出以赝电容为主的动力学特征、优异的倍率性能(10 A g⁻¹下225 mAh g⁻¹)以及卓越的循环耐久性。组装的全电池SHC器件在23 W kg⁻¹功率密度下实现了165 Wh kg⁻¹的高能量密度,在9424 W kg⁻¹的超高功率密度下仍保持108 Wh kg⁻¹,并在10,000次循环后容量保持率达到90%。该研究确立了通过设计异核位点以同时利用其固有功能异质性与电子离域效应,是克服储能材料动力学限制的有效策略。
在电化学储能领域,钠离子混合电容器(SHCs)通过协同集成电池的高能量密度与超级电容器的卓越功率密度和长循环寿命,展现出巨大应用前景。这种独特的结构使其成为需要快速充放电能力和持续能量输出的应用的理想选择,例如电网规模储能、再生制动系统和电动汽车。此外,由于钠资源的天然丰度和广泛分布,SHCs相较于锂基对应物(尤其是大规模应用)更具成本效益。尽管有这些优点,其广泛应用仍受制于一个核心的动力学瓶颈:负极中缓慢的Na⁺扩散和电荷转移,特别是在高电流密度下。法拉第负极与电容性阴极之间这种深刻的动力学失配会损害器件的倍率性能和循环稳定性,限制了最优能量-功率权衡。因此,理性设计具有本征加速离子传输的新型负极材料势在必行。
氮掺杂碳(N-C)材料已成为Na⁺存储的基础平台,具有可调的活性位点和改善的电导率。然而,其性能常受限于本征氮位点的低容量和中等吸附强度,而局部化的Na⁺-碳主体相互作用可能引发高扩散势垒和循环诱导的结构退化。为解决这一问题,开发了在N-C基质中原子级分散过渡金属(例如Fe、Co、Ni、Cu、Zn、Mn等)的单原子N-C材料。单原子最大限度地利用了金属原子,调控了局部电子结构,并创造了丰富的活性位点,从而增强了Na⁺的吸附和传输。例如,锚定在分级多孔N-C中的Fe单原子得益于丰富的活性位点和优化的Na⁺传输路径,在0.2 A g⁻¹的电流密度下循环1000次后,提供了249.4 mAh g⁻¹的可逆容量。N-C上负载的Co单原子(负载量极低,仅为0.12 wt.%)显著提升了Na⁺存储性能,在1.0 A g⁻¹的电流密度下循环2000次后容量达到285 mAh g⁻¹。此外,Cu单原子能够调控石墨相氮化碳中的N构型,在0.05 A g⁻¹电流密度下实现了350.4 mAh g⁻¹的比容量,并展现出卓越的倍率性能。然而,单金属单原子负极从根本上受限于其d轨道电子态的固有局域化。这种局域化常常导致过强的Na⁺吸附势阱,引发由Sabatier原理主导的经典"过强结合"限制:位点结合离子过强以致无法实现快速扩散,未能完全解决核心动力学挑战。因此,最大化性能需要一种新的设计范式,能够将Na⁺存储的热力学稳定性与扩散的动力学能垒解耦。
为了克服局域化单原子位点的限制,研究人员探索了双原子材料。这一新兴范式利用相邻(通常是异核)金属原子之间的协同电子相互作用,以解锁单原子材料所不具备的特性。例如,锚定在N-C上的Fe–Mn位点通过调控d带中心来优化氧中间体的结合,展现出卓越的双功能氧电催化活性。同样地,聚合物氮化碳上的Ni–Zn位点通过协同降低水氧化和氧还原的活化能垒,并借助电荷重分布以及电子和空穴的空间分离来稳定关键中间体(*OH和*OOH),从而显著提升了H₂O₂的光催化合成效率。这些发现共同强调了双原子可以理性调控电荷吸附和电荷转移动力学的原理,这对于加速Na⁺存储动力学同样至关重要。具体而言,打破动力学极限的两个关键要求定义如下:缓冲电荷并调节结合强度的电子机制,以及引导离子扩散的物理路径。
基于此原理,该研究策略性地配对Fe和Co以耦合其独特的电子结构。FeN₄位点具有较高的d带中心,会过度增强Na⁺结合,而CoN₄位点则具有较低的d带中心以提供中等强度的吸附。异核FeCo对本质上构建了一个由功能异质活性位点(FDASs)实现的捕获-释放动力学模型,其中结合较强的Fe位点实现高效的Na⁺捕获,而相邻的Co位点则促进低能垒的Na⁺迁移/脱附。同时,诱导强烈的d-d轨道耦合促成了轨道介导的电荷离域(OMCD)效应。在耦合d轨道与载体p轨道杂化的驱动下,轨道介导的电荷离域效应在双金属中心上创建了一个高度离域的电子系统,这对于调节能量景观以及降低Na⁺在这些功能异质活性位点之间迁移的动力学势垒至关重要。此外,离域电子云作为电荷缓冲层,增强了钠化/脱钠过程中的电荷补偿,并促进了表面诱导的赝电容,直接缓解了传统负极材料的动力学瓶颈。然而,这种双机制协同作用如何从量子力学原理上增强Na⁺扩散动力学尚待深入探索。因此,该研究通过构建模型FeCoN₆双原子位点(FeCo-N-C)来论证此原理。结合实验与理论的方法为OMCD效应与FDASs之间的协同作用提供了直接证据。至关重要的是,该研究清晰地阐明了这两个共存因素与显著增强的Na⁺存储动力学及卓越的长期稳定性之间的关联。该研究不仅展示了一种用于SHCs的高性能负极,还通过理性设计轨道层面的相互作用,为设计先进储能材料确立了新范式。
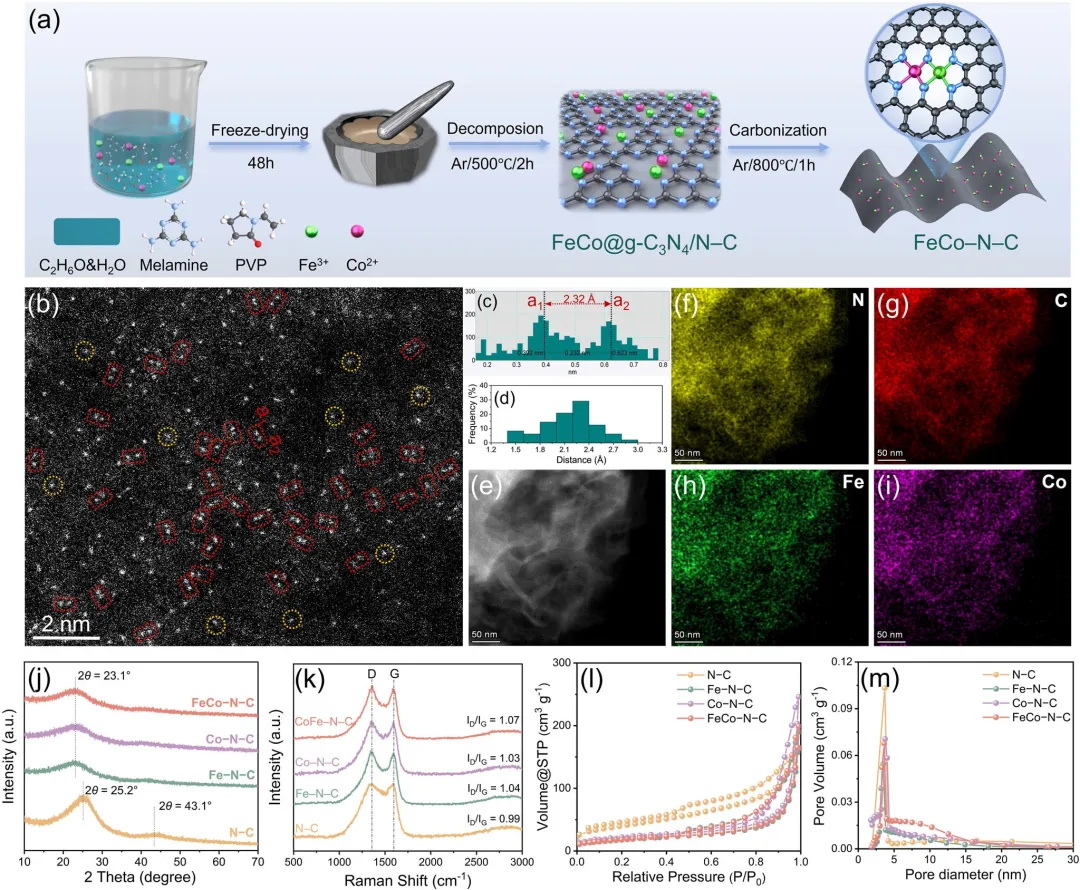
图1 | (a) FeCo–N–C的合成路线示意图。(b) FeCo–N–C的像差校正高角环形暗场扫描透射电子显微镜图像。(c) 一个代表性Fe–Co原子对的间距强度分布测量结果,以及(d) 根据像差校正高角环形暗场扫描透射电子显微镜图像测得的原子间距离统计分析(样本量:n = 40个原子对)。(e-i) FeCo–N–C的像差校正高角环形暗场扫描透射电子显微镜图像及其相应的元素分布图。(j) N–C、Fe–N–C、Co–N–C和FeCo–N–C的X射线衍射图谱,(k) 拉曼光谱,(l) N₂吸附-脱附等温线,以及(m) 相应的孔径分布。
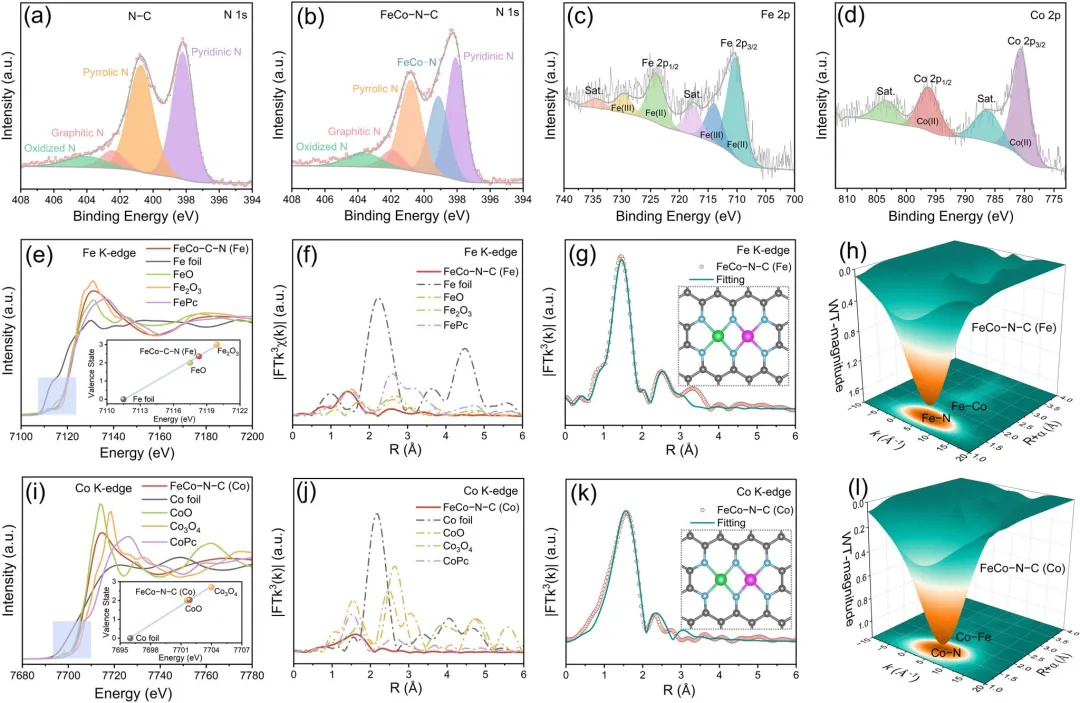
图2 | (a) N-C和(b) FeCo-N-C的高分辨率N 1s XPS谱。(c) Fe 2p和(d) Co 2p的高分辨率XPS谱(针对FeCo-N-C)。(e) Fe K边XANES谱(插图:基于吸收边能量位置的Fe氧化态线性拟合)及(f) FeCo-N-C的相应FT-EXAFS谱与参考材料(Fe箔、FeO、Fe₂O₃和FePc)对比。(g) Fe第一配位壳层的EXAFS拟合曲线(插图:提出的FeCo₆原子构型)及(h) FeCo-N-C中Fe位点的WT-EXAFS信号。(i) Co K边XANES谱(插图:基于吸收边能量位置的Co氧化态线性拟合)及(j) FeCo-N-C的相应FT-EXAFS谱与参考材料(Co箔、CoO、Co₃O₄和CoPc)对比。(k) Co第一配位壳层的EXAFS拟合曲线(插图:FeCoN₆原子构型)及(l) FeCo-N-C中Co位点的WT-EXAFS信号。
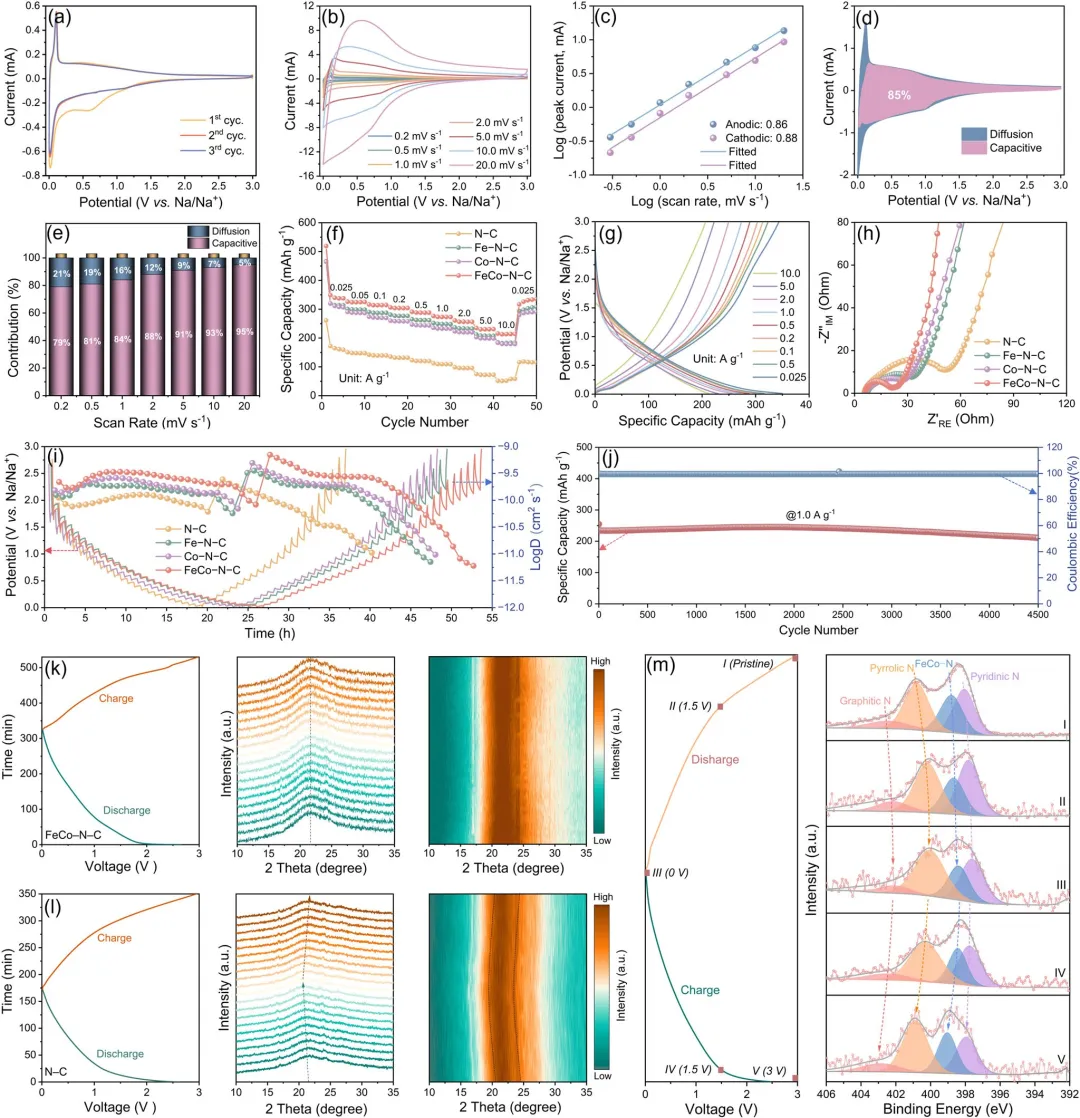
图3 | (a) 循环伏安曲线(前三个循环),(b) 不同扫描速率下的循环伏安曲线,(c) 峰值电流与扫描速率的依赖关系,(d) FeCo–N–C负极在1.0 mV s⁻¹扫描速率下的电容贡献占比(紫色区域),以及(e) FeCo–N–C负极在不同扫描速率下的电容贡献百分比柱状图。(f) N–C、Fe–N–C、Co–N–C和FeCo–N–C负极的倍率性能。(g) FeCo–N–C负极在不同电流密度下的恒电流充放电曲线。(h) N–C、Fe–N–C、Co–N–C和FeCo–N–C负极的电化学阻抗谱(EIS)奈奎斯特图及(i) 恒电流间歇滴定技术(GITT)曲线。(j) FeCo–N–C负极在1.0 A g⁻¹电流密度下的长期循环性能。原位XRD分析:(k) FeCo–N–C负极和(l) N–C负极(左:充放电曲线;中:XRD图谱;右:XRD的二维等高线图)。(m) 不同充放电状态下的非原位XPS N 1s谱(左:充放电曲线及采样点;右:对应于状态I-V的N 1s拟合谱)。
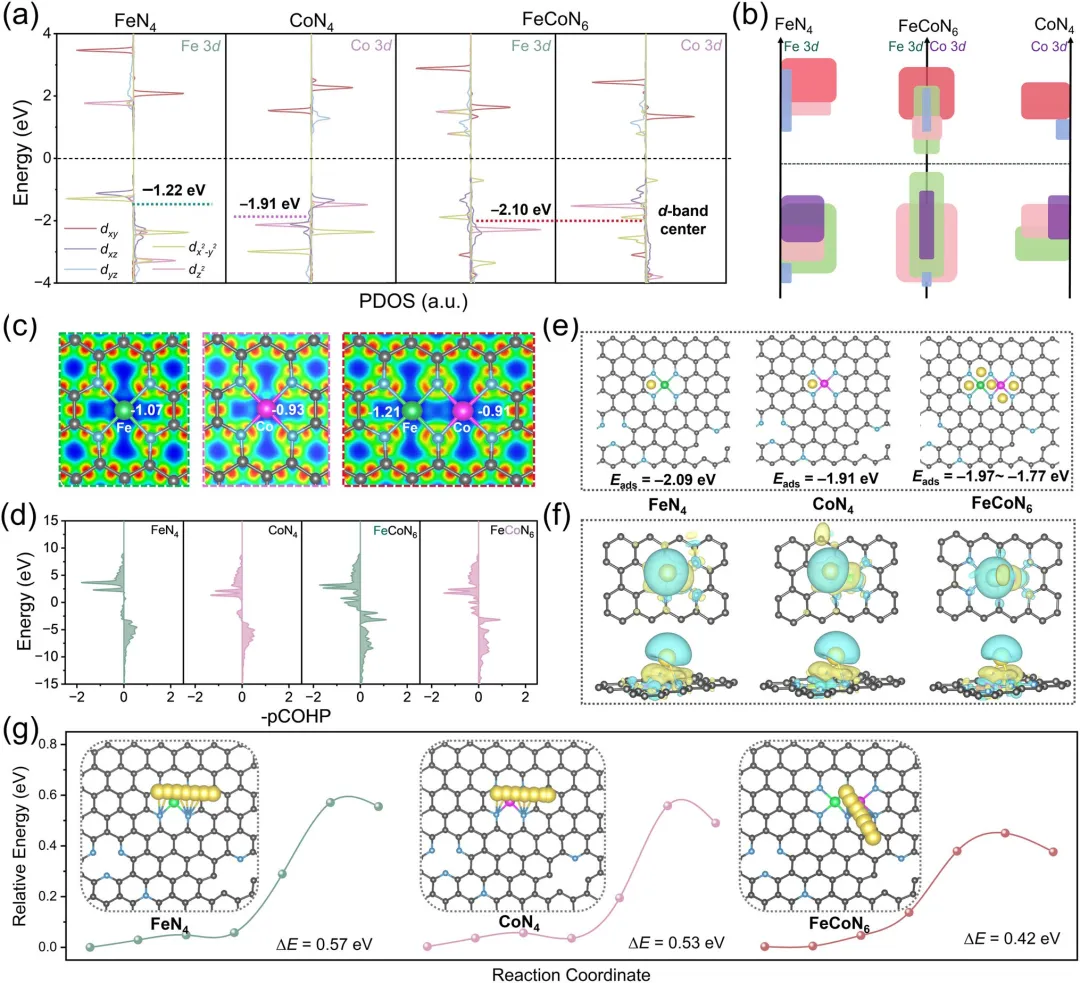
图4 | (a) Fe-N-C、Co-N-C和FeCo-N-C中Fe 3d和Co 3d轨道的投影态密度PDOS。(b) Fe 3d与Co 3d轨道相互作用的示意图。(c) 差分电荷密度及Bader电荷分析。(d) FeN₄、CoN₄和FeCoN₆中Fe-N和Co-N键的晶体轨道哈密顿布居分析。(e) Na⁺吸附的优化计算模型(FeN₄、CoN₄和FeCoN₆位点)及相应的吸附能。(f) 三维差分电荷密度等值面图(顶视图和侧视图)。(g) 不同表面Na⁺沿反应坐标扩散的相对能量分布及活化能垒。
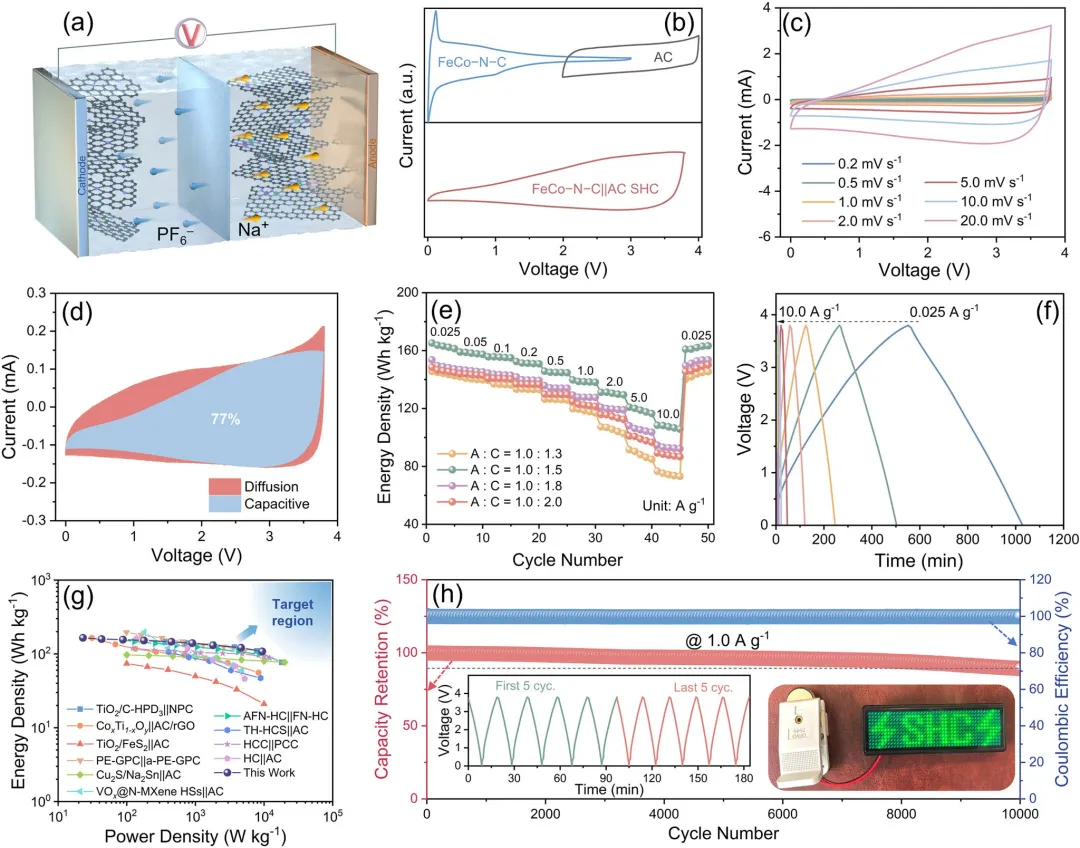
图5 | (a) FeCo-N-C||AC SHC器件示意图。(b) FeCo-N-C负极、AC正极及FeCo-N-C||AC SHC的电化学窗口(CV曲线)。(c) 不同扫描速率下的CV曲线。(d) 在1.0 mV s⁻¹扫描速率下,电容性与扩散控制贡献占比分析。(e) 不同阳极-阴极质量比下FeCo-N-C||AC SHC的能量密度。(f) 不同电流密度下的恒电流充放电曲线。(g) 能量-功率特性对比图。(h) FeCo-N-C||AC SHC在1.0 A g⁻¹下的长期循环性能。插图:最初与最后5个循环的电压曲线,以及器件点亮LED组成的"SHC"图案演示。
总之,该研究在异核FeCoN₆位点中确立了一种新颖的双机制协同效应:其固有的异核性质创造了用于协同捕获-释放路径的功能异质活性位点,同时强d-d轨道耦合诱导了电子重构,产生了OMCD效应。研究人员证实,该OMCD效应从电子层面调制了固有的功能异质活性位点,通过下移集体的d带中心来调节Na⁺结合亲和力,规避了单原子位点的过强结合动力学瓶颈,从而同时优化了热力学和动力学。同时,还降低了Na⁺沿此协同捕获-释放路径的扩散势垒(FeCoN₆:0.42 eV vs. FeN₄:0.57 eV 与 CoN₄:0.53 eV)。因此,优化后的FeCo–N–C负极表现出主导的赝电容动力学特性(1.0 mV s⁻¹下贡献率为85%)、高Na⁺扩散系数(5.1×10⁻¹¹ cm² s⁻¹)、优异的倍率性能(10.0 A g⁻¹下容量为225 mAh g⁻¹)以及卓越的长期稳定性(1.0 A g⁻¹下循环4,500次)。值得注意是,组装的FeCo–N–C||AC SHC器件展现出优异的实际指标:165 Wh kg⁻¹的高能量密度、9424 W kg⁻¹的超高功率密度(保持108 Wh kg⁻¹的能量密度)以及优异的耐久性(10,000次循环后容量保持率为90%)。该研究表明,通过设计异核位点以同时利用本征FDASs和OMCD效应,是理性设计下一代高性能储能材料的一种强有力策略。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
