相工程对于优化电催化剂的电子结构和反应路径至关重要,可在单相结构的高效电荷转移与混合相结构的协同效应之间取得平衡,然而控制相演化的基本原理仍不清楚。
2026年04月20日,深圳大学任祥忠、罗兆艳团队在Angewandte Chemie International Edition期刊发表题为“Unveiling the Valence-Driven Charge Compensation Mechanism to Direct Phase Engineering in Ru-Based Catalysts for Acidic Water Electrolysis”的研究论文,团队成员周戌燕为论文第一作者,任祥忠、罗兆艳为论文共同通讯作者。
第一作者:周戌燕
通讯作者:任祥忠、罗兆艳
通讯单位:深圳大学
论文DOI:10.1002/anie.7924702
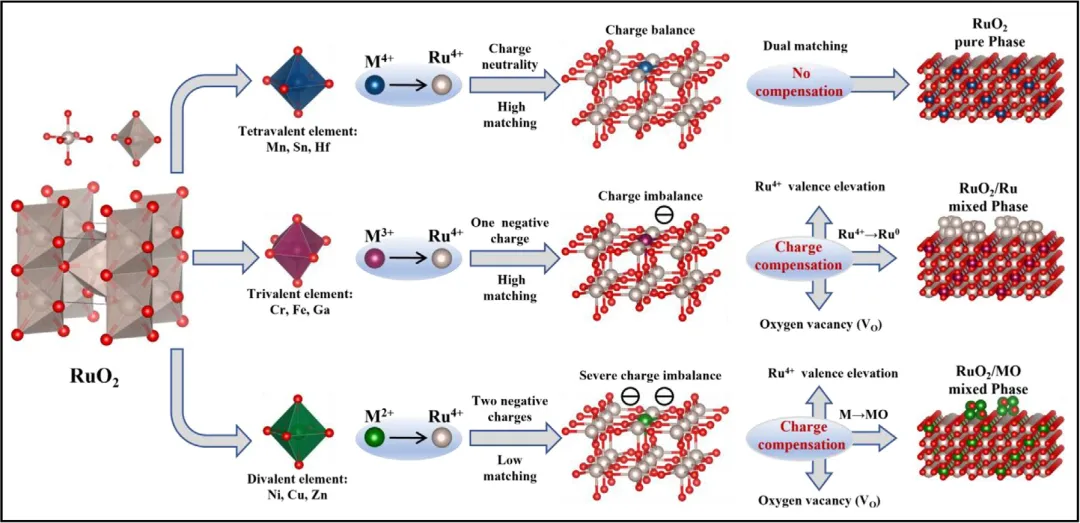
该研究建立了一种价态驱动的相工程范式,能够对钌基催化剂实现精确调控。该研究揭示,相形成轨迹本质上由电荷补偿机制决定:四价掺杂剂(Hf、Mn、Sn)通过电荷中性和晶格匹配维持单相RuO₂;三价掺杂剂(Cr、Fe、Ga)诱导产生RuO₂-Ru协同异相结构;而二价掺杂剂(Ni、Cu、Zn)由于严重的电荷失配引发氧化相分离。作为示例,RuGa混合相催化剂在10mA cm⁻²电流密度下表现出180mV的优异过电位,并在酸性介质中稳定运行500小时。当集成到质子交换膜水电解槽中时,该催化剂在仅1.63V电压下即可达到1A cm⁻²的电流密度,并在500mA cm⁻²下具有100小时的稳健稳定性。该研究为通过价态调控的相调节策略合理设计耐用的酸性OER催化剂提供了基本框架。
质子交换膜水电解(PEMWE)技术凭借其快速响应动力学、高操作电流密度和优异的气体纯度,处于全球向绿色氢能经济转型的前沿。尽管有这些优势,PEMWE的大规模部署仍受到阳极OER的严重制约,该反应不仅动力学缓慢,而且需要使用贵金属催化剂来耐受腐蚀性酸性环境。虽然铱基氧化物是目前的事实标准,但其稀缺性和高昂成本迫使人们开发更丰富的替代材料。金红石相RuO₂因其优异的本征活性和较低成本而成为最有前景的候选材料;然而,其实际应用受到高氧化电位下剧烈溶解的阻碍,这一过程与晶格Ru位点的过度氧化及随后的结构坍塌本征相关。
为了弥合活性与稳定性之间的差距,结构工程——特别是相调控——已成为优化钌活性位点的有力工具。当前研究通常分为两种范式:构建单相系统(例如通过杂金属掺杂形成固溶体)以调控钌活性位点的电子结构和配位环境,从而提升催化性能;或者制造多相系统(例如异质结或负载型催化剂),利用界面相互作用促进协同催化效应。虽然这两种方法都取得了优异性能指标,但其背后的设计规则仍然是现象性的。关键的是,掺杂剂价态与相演化轨迹之间的基本关联尚未得到系统阐明。对于如何通过掺杂剂诱导的晶格扰动触发的电荷补偿机制进行策略性操控以指导特定相的形成,目前仍缺乏理解。
在此,该研究提出一种价态驱动的电荷补偿机制来弥合这一知识空白,该机制实现了对钌基催化剂相结构从多相复合物到单相金红石结构的精确控制。通过密度泛函理论DFT计算与实验结构表征的协同结合,该研究揭示相结构本质上由掺杂剂价态所决定的特定电荷补偿路径所支配。具体而言,研究人员证明三价Ga掺杂的RuO₂体系演变为一种独特的RuO₂-Ru混合相结构,该结构优化了活性位点密度与结构完整性之间的平衡。所得到的催化剂在10mA cm⁻²电流密度下仅表现出180mV的优异过电位,并具有超过500小时的稳定性。在实际PEMWE评估中,该催化剂在1A cm⁻²下实现1.63V的电池电压,并在500mA cm⁻²下持续稳定运行100小时。该研究建立了一个调控贵金属催化剂相景观的综合范式,为经济耐用型PEMWE系统铺平了道路。
图1 | 四价、三价、二价金属掺杂的价态驱动相调控机制。
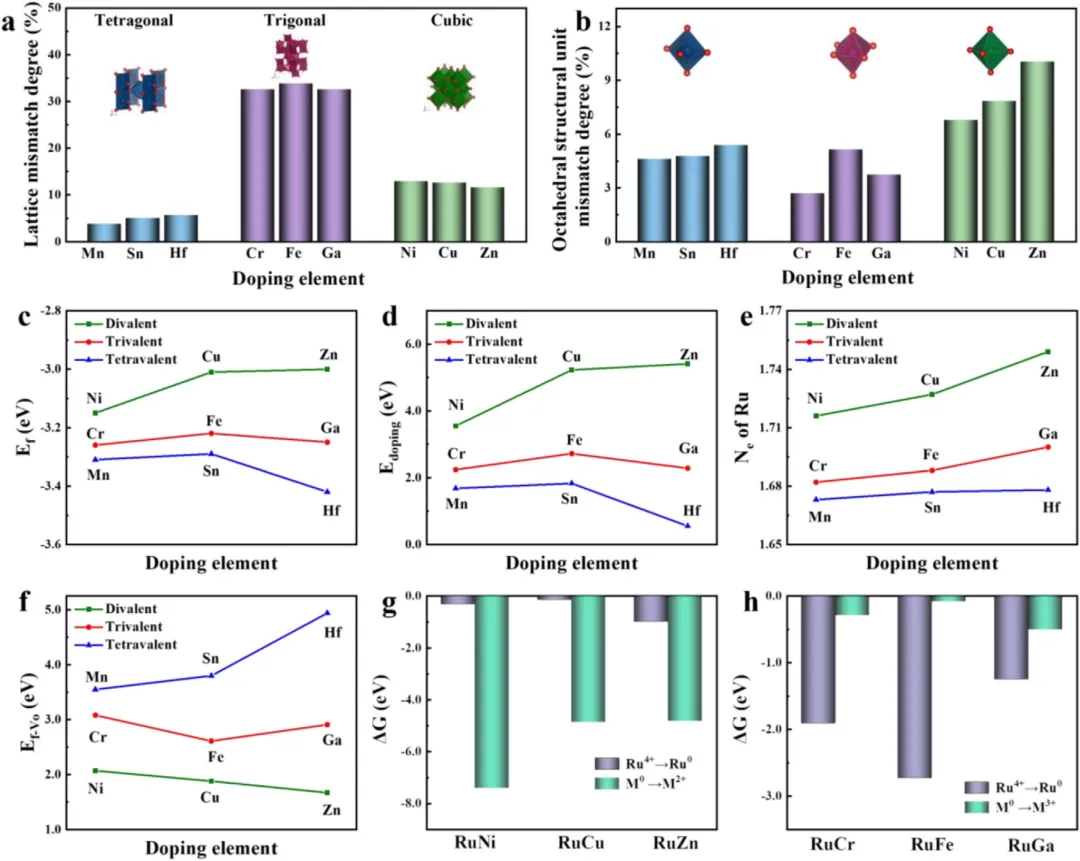
图2 | 掺杂元素氧化物与RuO₂之间的晶格失配度(a)和八面体结构失配度(b)。RuM催化剂的计算形成能(c)和掺杂能(d)。Ru损失的电子数(e)。RuM催化剂的氧空位形成能(f)。二价金属掺杂时Ru⁴⁺→Ru⁰和M⁰→M²⁺的吉布斯自由能(ΔG)计算(g)。三价金属掺杂时Ru⁴⁺→Ru⁰和M⁰→M³⁺的吉布斯自由能(ΔG)(h)计算。
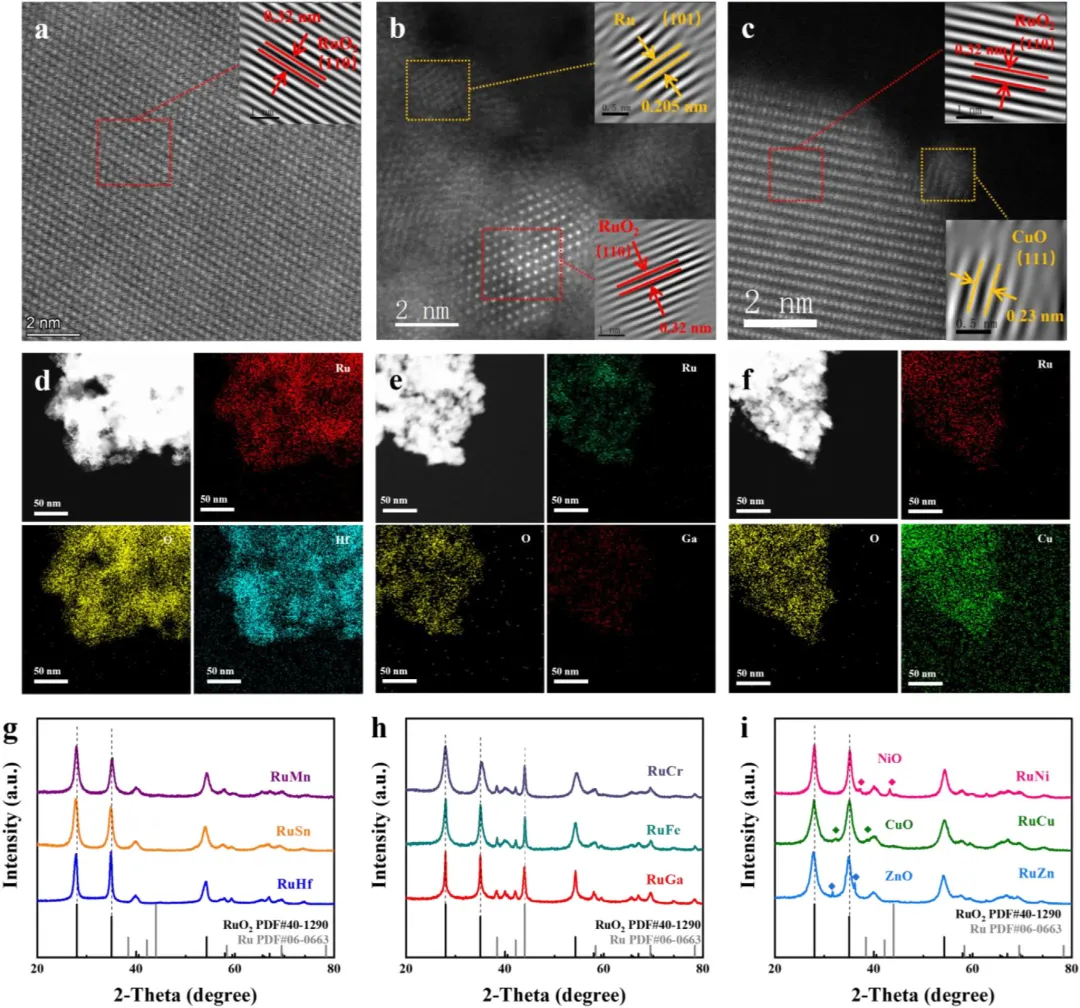
图3 | 结构与组分表征。RuHf(a)、RuGa(b)和RuCu(c)的HAADF-STEM图像。RuHf(d)、RuGa(e)和RuCu(f)的EDS元素分布图。四价(g)、三价(h)和二价(i)掺杂的XRD图谱。
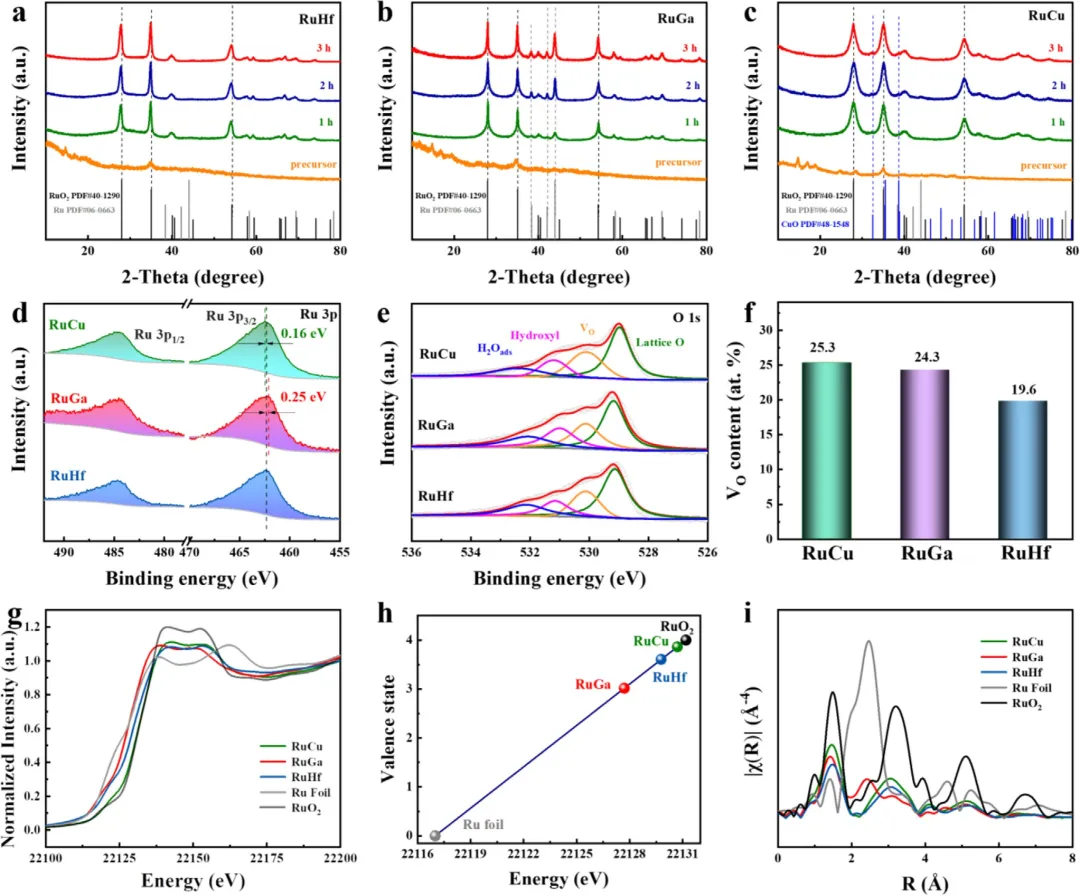
图4 | 各样品的结构与组分表征。RuHf(a)、RuGa(b)和RuCu(c)在退火过程中的XRD图谱。(d) RuHf、RuGa和RuCu的高分辨XPS Ru 3p谱图。(e) RuHf、RuGa和RuCu的高分辨XPS O 1s谱图。(f) RuHf、RuGa和RuCu的氧空位比例。(g) RuHf、RuGa、RuCu及参考样品的Ru K-edge XANES谱图。(h) RuM及参考样品的Ru K-edge吸收能与氧化态之间的线性关系。(i) RuHf、RuGa、RuCu及参考样品的Ru K-edge FT-EXAFS谱图。
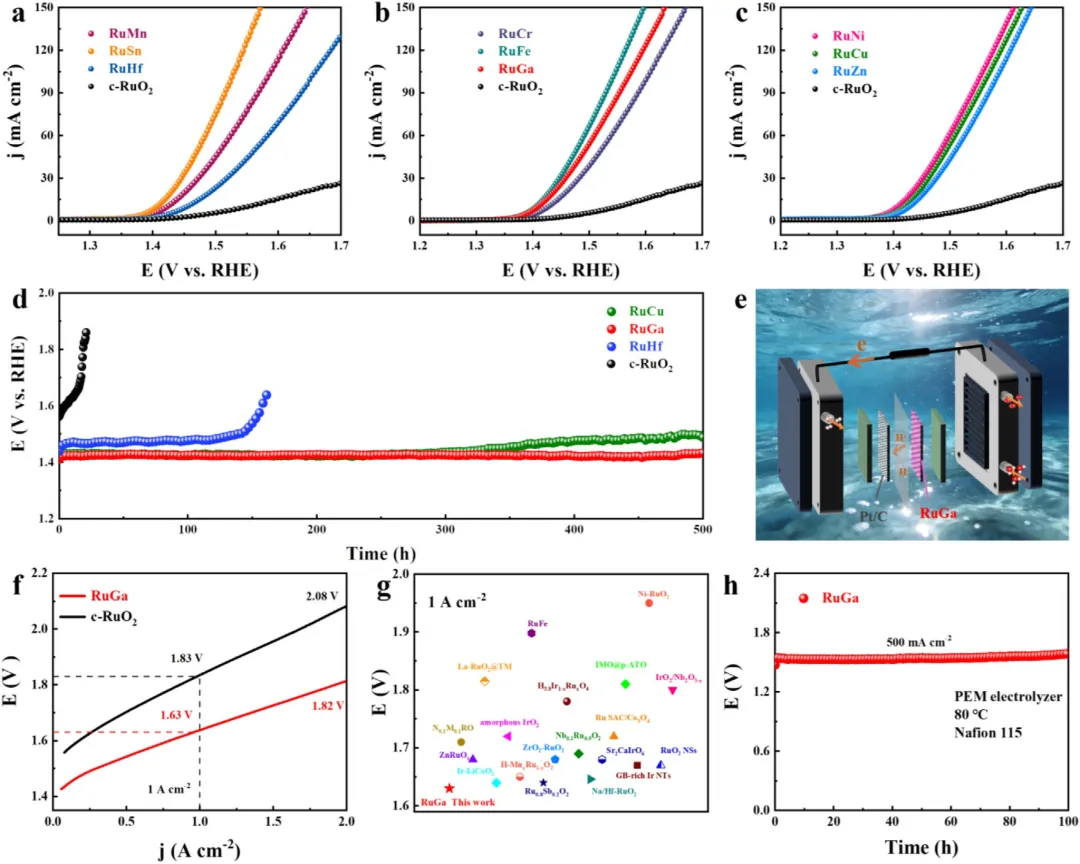
图5 | 在O₂饱和的0.5M H₂SO₄溶液中,四价(a)、三价(b)和二价金属掺杂(c)的OER极化曲线。(d) RuHf、RuGa、RuCu及商业RuO₂在10mA cm⁻²电流密度下的计时电位测试。(e) PEM电解槽示意图。(f) 以RuGa和商业RuO₂为阳极电催化剂、采用Nafion 115膜在80°C下测得的极化曲线。(g) 在1A cm⁻²下的电池电压与文献数据的比较。(h) 采用RuGa催化剂、在500mA cm⁻²和80°C下、使用Nafion 115膜的计时电位曲线。
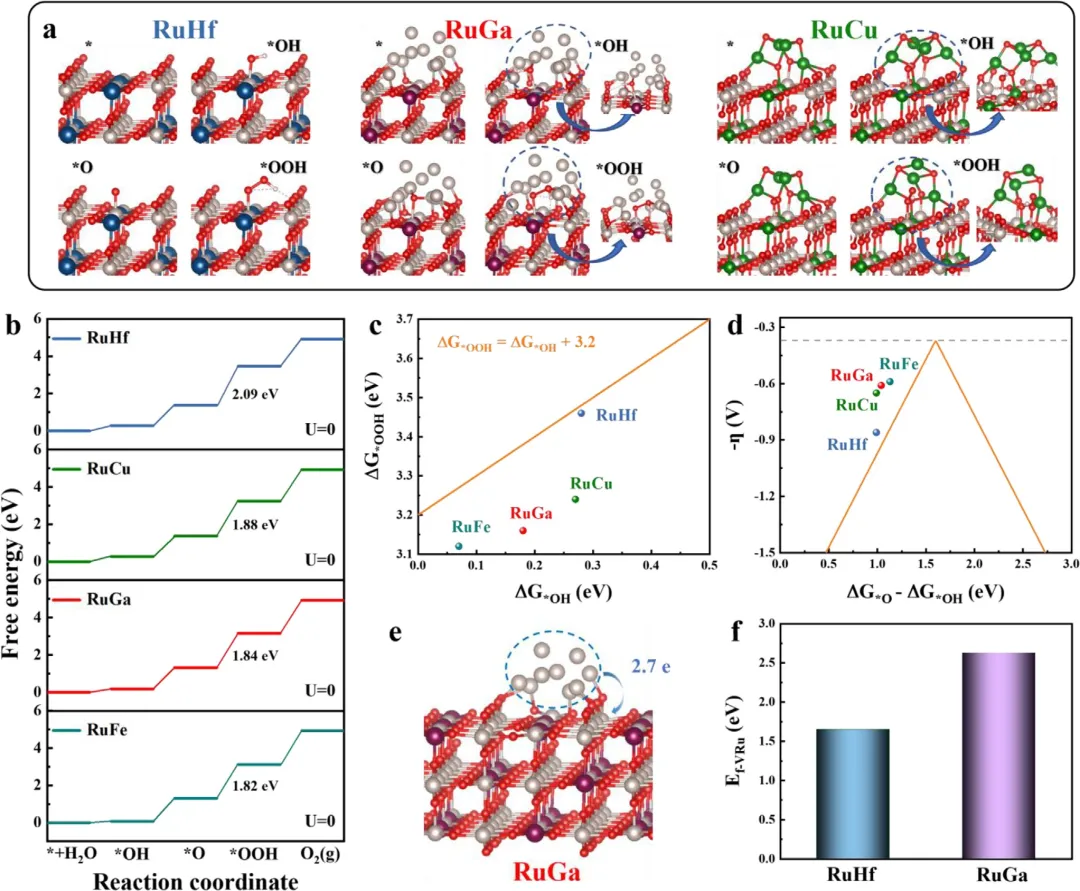
图6 | (a) RuHf、RuGa和RuCu的三种反应中间体(*OH、*O和*OOH)的最终结构。(b) RuHf、RuGa、RuCu和RuFe的OER自由能图。(c) *OH和*OOH物种吸附自由能之间的线性关系。(d) OER理论过电位(η)与(ΔG*O - ΔG*OH)的火山图。(e) RuGa中Ru簇向基底损失的电子数。(f) RuHf和RuGa的Ru空位形成能(Ef)。
总之,该研究提出了一种价态驱动的相工程策略,通过调控掺杂金属的价态,实现了对钌基OER催化剂相结构的精确控制。其核心机制涉及三种不同的路径:四价掺杂剂通过电荷中性和晶格兼容性促进单相RuO₂的形成;三价掺杂剂通过多步电荷补偿驱动体系形成RuO₂-Ru混合相;而二价掺杂剂由于严重的电荷失配和晶格破坏导致RuO₂-MO相分离。该机制通过集成的密度泛函理论DFT计算和全面的结构表征得到了理论和实验验证。在所研究的催化剂中,RuGa混合相催化剂表现出优异的酸性OER性能,在10mA cm⁻²电流密度下仅需180mV的过电位,并具有超过500小时的稳定性。当集成到PEMWE槽中时,RuGa在1A cm⁻²下提供1.63V的电池电压,并在500mA cm⁻²下维持稳定运行100小时,超越了商业RuO₂,并与最先进的催化剂相比具有竞争力。该研究建立了一个通用的价态调控设计框架,用于Ru基酸性OER催化剂,为实现高效耐用的绿氢生产提供了一条理性途径。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
