顶刊速报|深圳大学彭孝军院士、中国科学院化学所李玉良院士和北京化工大学孙晓明教授等电催化剂最新成果速览
- 2026-06-19 17:54:05
引言
随着全球对清洁能源需求的日益增长,氢能作为高效、清洁的能源载体,其开发与利用对于实现“双碳”目标至关重要。以电解水为代表的电化学能源转换技术,是实现氢能高效转化的核心。然而,其规模化发展仍面临氢析出反应(HER)与氧析出反应(OER)动力学迟缓、催化剂成本过高及稳定性不足等关键瓶颈。在波动性可再生能源并网的条件下,电催化剂还需同时兼顾高活性、长寿命与高效率等严苛要求。
为突破这些瓶颈,近年来研究聚焦于通过原子级精准设计与多维结构调控策略,开发高性能、高稳定性、低贵金属用量的新型电催化剂。研究热点涵盖:通过杂原子精准掺杂尖晶石晶格兼顾酸性OER活性与PEMWE稳定性;发展石墨炔诱导的高自旋态过渡金属原子,打破自旋禁阻效应驱动高效酸性氧析出;建立双描述符理论框架,高通量筛选耐用的金属掺杂RuO2酸性OER催化剂。同时,小分子电氧化辅助节能制氢成为替代传统OER的变革性路径:界面工程赋能肼氧化(HzOR)双功能催化剂,实现低过电位制氢;Re、Ru共掺杂过渡金属合金驱动甘油电氧化高选择性合成甲酸酯并耦合产氢;磷掺杂诱导钴自旋态演化,通过原位X射线发射光谱解析乙二醇电氧化中C-C键选择性断裂机制;Ce-Mo双调控亚稳态Ni2P相变,实现高效尿素电解与析氢双功能。这些研究从电子自旋态、晶格配位环境、界面微结构及反应路径等多维度协同优化催化剂性能,推动其在质子交换膜水电解槽(PEMWE)及小分子辅助制氢集成器件中的应用。本篇汇总了深圳大学彭孝军院士、燕山大学张庆瑞教授、青岛科技大学王磊教授、中国科学院化学研究所李玉良院士、北京化工大学孙晓明教授、内蒙古大学武利民教授、安徽大学李鹏教授、哈尔滨工业大学于永生教授等课题组在电催化材料设计、机理探索与器件应用方面的最新突破性进展。
1. Nano Research:通过Cr3+精准掺杂Co3O4尖晶石八面体晶格兼顾质子交换膜电解槽活性与稳定性
IrO2是最常见的酸性OER催化剂,但由于贵金属元素稀有、成本过高等因素,严重限制了其在PEMWE领域的大规模应用。而Fe、Co、Ni等过渡金属基电催化剂凭借其丰富的外层d轨道电子和多变的氧化态成为极具潜力的替代品,尤其是具有独特的电子结构和催化活性的钴基尖晶石结构Co3O4。然而,目前,纯相Co3O4在酸性OER中的催化活性和稳定性还远未达到工业生产的要求。为解决这一问题,部分研究尝试通过杂原子掺杂或取代策略来引入氧空位并调控电子结构。Cr等VI-B族阳离子由于具有丰富的开放轨道,能有效改变Co的电子排布从而激发OER性能,在各种掺杂元素中尤为突出。但是现有Cr-Co复合氧化物催化剂在酸性介质中的过电位和稳定性问题仍未解决,且反应动力学较为迟缓,难以满足高电流密度运行需求。此外,尽管Cr掺杂能有效提升Co3O4中Co-O键的共价性并优化CO2+/Co3+比例,但Cr在晶格中的精准占位及其对酸性OER路径的调控机制仍不明确,阻碍了高性能催化剂的设计。
鉴于此,深圳大学彭孝军院士和内蒙古师范大学李杲教授等通过一步热解-空气退火策略成功制备了Cr3+精准占据八面体位点的CrCo8Ox尖晶石固溶体催化剂,得益于Cr3+掺杂诱导的富氧空位、Co3+/Co4+氧化对及优化的d带中心,该催化剂在酸性OER和PEMWE中同时展现出优异的活性与稳定性。这项工作为开发高效的PEMWE催化剂提供了重要参考。
本文要点:
1) 首先采用简单的一步硝酸盐热解-空气退火法成功制备了碳纸负载的尖晶石结构CrCo8Ox催化剂,Cr、Co、O元素均匀分布,Cr3+选择性取代八面体位的Co3+,形成稳定的固溶体;
2) X射线光电子能谱和拉曼光谱结果表明,Cr的引入显著提高了Co3+/Co2+比例并诱导产生大量氧空位,同时Cr3+以Cr-O键形式稳定嵌入晶格,增强了催化剂的结构刚性与抗氧化能力;
3) 在0.5 M H2SO4电解质中,CrCo8Ox催化剂在10 mA cm-2电流密度下的酸性OER过电位仅292 mV,Tafel斜率低至35 mV dec-1,优于商业IrO2,并能在100和300 mA cm-2下分别稳定运行130小时和20小时;
4) 将CrCo8Ox作为阳极催化剂组装PEMWE膜电极,在80℃下实现了优异的电解性能,可在100 mA cm-2下连续运行130小时,电压保持约1.9 V,展现出良好的工业应用潜力;
5) 原位红外光谱和密度泛函理论计算结果表明,Cr3+掺杂使Co3O4的反应路径从氧化物路径机制(OPM)转为吸附质演化机制(AEM),并通过下调自旋向上d带中心和上调自旋向下d带中心,显著降低*OOH中间体的形成能垒,从而协同提升了催化活性与稳定性。
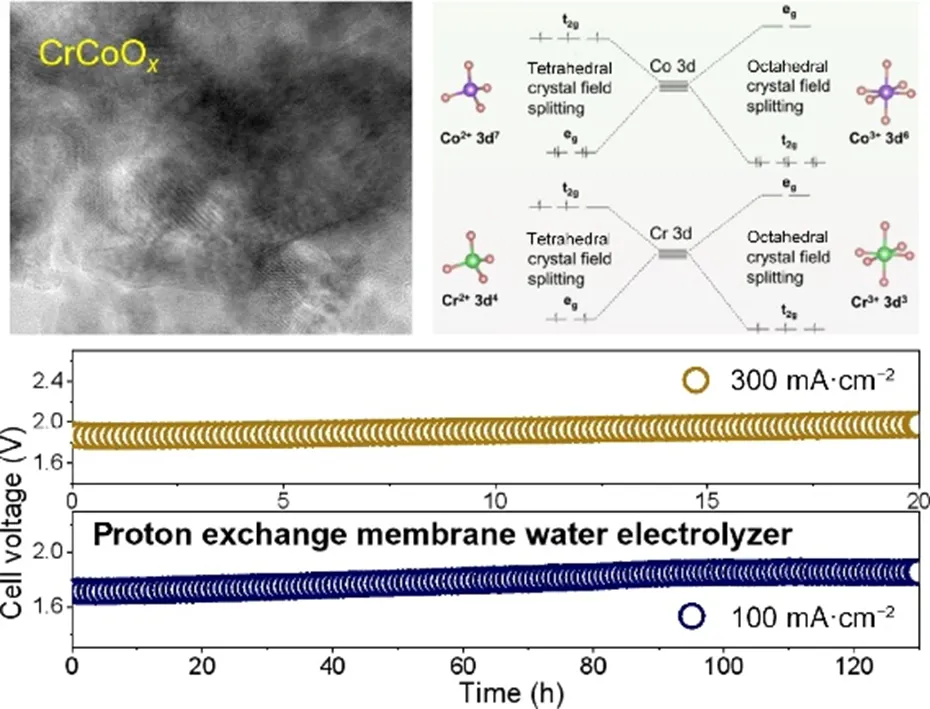
Piracha Sanwa, et al., Simultaneous activity and stability gains in PEM electrolyzer via targeted Cr3+ occupation in spinel Co3O4 octahedral lattices. Nano Res., doi:10.26599/NR.2026.94908636.

识别二维码访问原文
2. Nano Research综述:肼辅助制氢双功能电催化剂的界面工程
为克服OER理论电位高和动力学缓慢的固有局限性,研究者近年来尝试开发基于乙醇、葡萄糖、尿素和硫离子等小分子的替代氧化反应。其中,具有极低过电位的肼氧化反应(HzOR)脱颖而出,且仅产生唯一产物N2,在热力学电位和环境友好性方面具有显著优势。从高性能催化剂理性设计的角度出发,一方面,需要调控形貌以暴露更多活性位点,从而提升本征活性;另一方面,需要调控电子结构,控制电子转移过程,改善反应中间体的形成和最终产物的脱附过程。对此,可借助界面工程有效调控催化剂的形貌和结构。基于界面工程设计的催化剂通常包含两个或多个组分,通过合理调控界面处的原子排列,可有效调控电催化剂的物理化学性质。此外,不同组分之间的强耦合相互作用以及工程化的界面结构,能够提升电子传导效率。近年来,电催化剂研究取得了显著进展,但缺少针对肼氧化辅助制氢体系中双功能催化剂的系统性综述。
鉴于此,燕山大学张庆瑞教授、王静教授、内蒙古工业大学赵二俊教授和巴基斯坦拉合尔管理科学大学Ali auf博士等系统总结了用于肼辅助水分解的双功能电催化剂的界面工程策略,重点阐述了HER和HzOR的反应机理与关键描述符,详细介绍了界面结构解析的表征技术,并深入讨论了空位缺陷、掺杂调控、异质结构、应变工程和单原子催化等策略在优化催化性能方面的作用。这项工作为设计高效、节能、稳定的双功能HzOR辅助制氢催化剂提供了系统的理论指导。
本文要点:
1) 首先系统阐述了碱性介质中HER的Volmer-Heyrovsky/Tafel机理及HzOR的逐步脱氢机理,指出HER的决速步为Volmer反应(水分解),而HzOR的决速步为N2Hx中间体的脱氢过程,并强调氢吸附自由能(ΔG*H)作为关键描述符在火山图评价催化剂本征活性中的核心作用;
2) 总结了高分辨透射电镜、高角环形暗场扫描透射电镜、X射线光电子能谱、X射线吸收谱以及原位拉曼/原位X射线吸收谱等表征技术在揭示界面原子结构、电子转移、配位环境及反应中间体动态演化中的独特优势;
3) 系统归纳了界面工程策略,包括阴离子/阳离子空位缺陷通过调控电子结构和降低反应能垒提升HER/HzOR性能,非金属和金属掺杂及阴阳离子共掺杂通过协同优化中间体吸附能和高指数晶面暴露增强催化活性,异质结构利用界面内建电场促进水分解和氢溢流,应变工程通过晶格失配诱导压缩或拉伸应变调控d带中心位置,以及单原子催化剂利用最大原子利用率和金属-载体界面协同效应显著提升HzOR和HER性能;
4) 通过对比多种界面调控策略下的催化剂性能,指出在1.0 M KOH/0.5 M N2H4电解液中,部分优化催化剂表现出超强性能,然而在工业级电流密度下的长期稳定性、反应机理的原位可视化解析以及机器学习辅助高效筛选最优界面结构等方面仍面临挑战,未来应致力于拓展耦合生物质转化、塑料降解等创新集成体系。
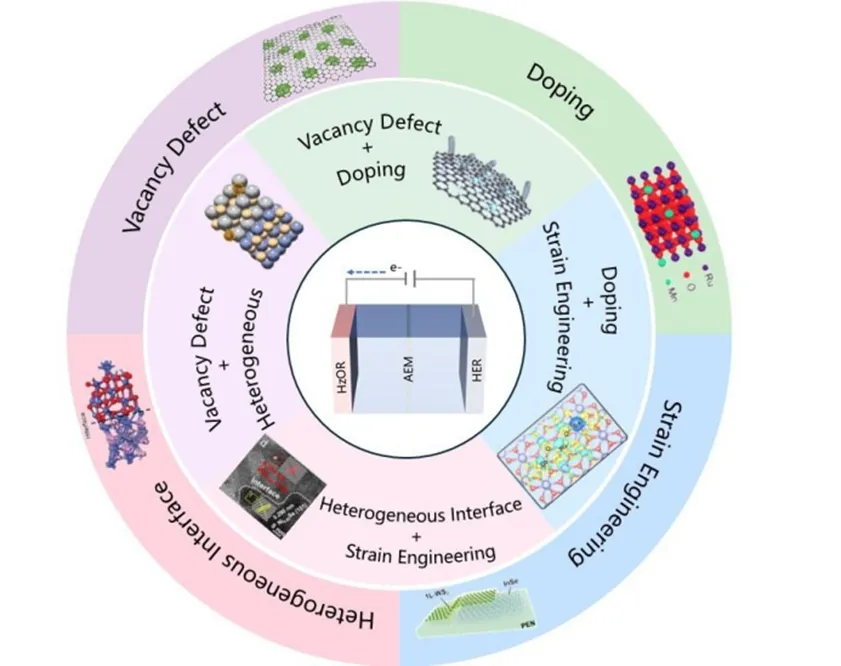
Wenxin Wang, et al., Interface engineering of bifunctional electrocatalysts for hydrazine-assisted hydrogen production. Nano Res., doi:10.26599/NR.2026.94908709.

识别二维码访问原文
3. Nano Research综述:小分子电氧化辅助节能制氢电催化剂的设计原则
近年来,小分子电氧化反应(SMOR)在电解水制氢领域成为替代传统OER的研究热点,SMOR不仅在热力学上更具优势,在工业生产中还可显著降低制氢能耗。然而,目前SMOR辅助制氢体系仍面临催化剂中毒、多反应路径竞争以及高电流密度下传质受限等严峻挑战。例如,Pt基催化剂在甲酸氧化中因CO吸附可在30分钟内损失超过40%的活性;葡萄糖电氧化中C1与C6路径的选择性调控要求精确的电子结构设计。为解决这些难题,研究者尝试了多种策略,包括维度调控、表面化学修饰和电子传输路径优化。然而,尽管HzOR、硫氧化(SOR)、尿素(UOR)氧化、醇氧化等各类SMOR研究成果层出不穷,但系统总结其电催化剂通用设计原则的综述仍付之阙如。现有综述多聚焦于单一反应类型或特定材料体系,缺乏对活性位点、界面工程、电子转移路径等跨反应普适规律的统一框架。难以从碎片化的信息中将单一反应中习得的设计智慧迁移应用到另一种反应。因此,亟需建立一个系统性的设计原则体系,以指导高效、稳定、低成本的SMOR电催化剂开发,从而发挥其节能潜力。
鉴于此,青岛科技大学王磊教授和刘晓斌副教授等人系统综述了SMOR辅助制氢电催化剂的设计原则,重点从热力学优化、催化剂维度调控、表面化学工程以及电子传输路径四个维度,全面阐述了提升SMOR催化性能的核心策略,并深入剖析了当前面临的稳定性、选择性及工业化放大等关键挑战。这项工作为开发高效节能的SMOR辅助制氢体系提供了系统的理论指导和未来研究方向。
本文要点:
1) 系统阐述了SOR、HzOR、UOR、乙醇氧化(AOR)、乙二醇氧化(EGOR)、甲醇氧化(MOR)等SMOR替代OER的热力学优势与动力学瓶颈,指出其理论电位极低,可在0.3-0.5 V的超低槽压下实现产氢,能耗较传统电解水降低30%-60%;
2) 归纳了0D单原子/团簇、1D纳米线/纳米管、2D纳米片、3D纳米阵列/多孔框架等催化剂维度调控策略对活性位点暴露、电子结构及传质动力学的影响,强调分级结构设计可同时提升反应速率与稳定性;
3) 总结了异质结构建、元素掺杂、缺陷工程、合金化和晶相调控等可有效调控d带中心与中间体吸附能的表面化学精准调控方法;
4) 讨论了电子传输路径优化途径,通过构建Ru-O-M桥键、高导电载体和三维自支撑电极,降低电荷转移电阻,实现在安培级电流密度下的长时间稳定运行;
5) 最后面向工业应用,提出了未来研究重点:发展抗中毒策略、结合原位表征与DFT计算揭示反应机理、开发可规模化制备的高电流密度催化剂以及解决大尺寸电极的均一传质与电场分布问题。
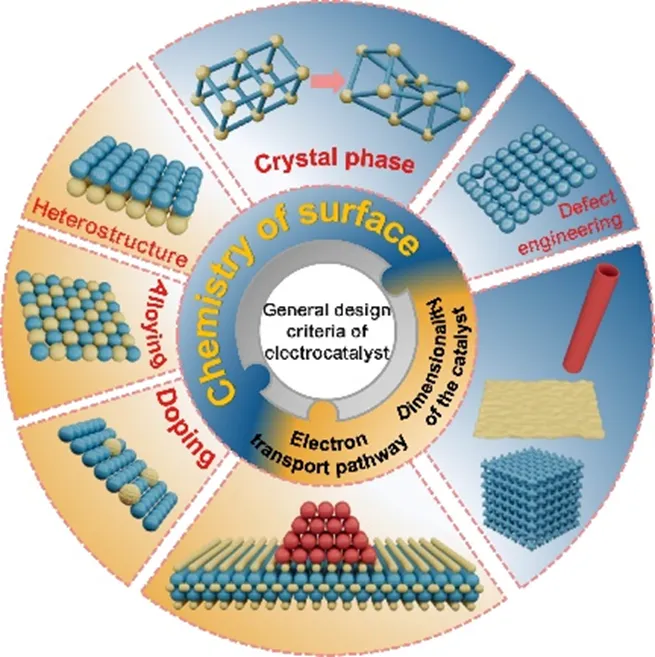
Zefeng Teng, et al., Design principles of electrocatalysts for energy-saving hydrogen production assisted by electrochemical oxidation of small molecules, Nano Res., doi:10.26599/NR.2026.94908237.

识别二维码访问原文
4. Nature Communications:高自旋过渡金属原子驱动酸性析氧电催化
过渡金属基催化剂的催化性能根本上受限于含氧中间体的自旋禁阻效应——为满足角动量守恒,生成单线态激发态O2需要额外约1.0 eV的能量。一些研究尝试通过自旋态调控来调节催化活性中心的d轨道占据数,适当的自旋构型可借助洪特规则驱动的平行自旋排列降低O-O耦合能垒,然而在众多非贵金属体系中,通过自旋态控制同时优化水解离和氧二聚化的清晰路线仍不明晰。经过多年探索,具有成本、储量和OER活性优势的Co3+从众多过渡金属候选材料中脱颖而出。不过传统Co基催化剂设计策略主要聚焦于电子结构调控而忽略了自旋态工程,而通过提升Co3+的自旋态可有效激活其eg轨道,从而增强OER动力学。尽管如此,部分已被报道的高自旋Co3+基氧化物在酸性PEMWE条件下表现出极不稳定、迅速降解的现象。对此,可以尝试开发能在界面产生强金属-载体相互作用并保持高催化性能的耐腐蚀催化体系以解决这一问题。作为一种新型碳材料,石墨炔(GDY)拥有独特的sp/sp2杂化碳骨架、本征纳米孔结构、高电子迁移率以及强金属锚定能力,但其是否能够通过金属-碳键合调控金属原子的d轨道占据数并诱发自旋态跃迁,进而优化OER性能,仍有待深入探索。
鉴于此,中国科学院化学研究所李玉良院士和吉林大学薛玉瑞教授等人利用石墨炔诱导钴基氧化物(HSS-CoOx/GDY)产生高自旋态Co3+,通过调控CoO6八面体的Jahn-Teller效应和晶体场分裂能,打破了自旋禁阻效应并优化了含氧中间体的吸附/脱附行为,从而在酸性OER中展现出优异的催化活性和稳定性。这项工作表明了石墨二炔在操纵电催化剂电子自旋态方面的能力。
本文要点:
1) 密度泛函理论计算预测,GDY的富电子π共轭体系与Co原子键合可诱发CoO6八面体的Jahn-Teller效应,降低晶体场分裂能,促进t2g电子向eg轨道跃迁,从而使Co3+从低自旋态转变为高自旋态,高自旋中心将决速步能垒降低;
2) 采用脉冲电沉积结合低温热氧化法制备了三维多孔网络结构HSS-CoOx/GDY厚纳米片,Co3O4纳米颗粒在GDY表面均匀分散,并发生晶格畸变和结构缺陷,产生了大量阶梯型界面;
3) 拉曼光谱、X射线光电子能谱和X射线吸收谱结果表明,石墨炔与Co3O4之间形成了sp-C-Co键并破坏了CoO6八面体的结构对称性,导致Co-O键长伸长,Co的氧化态降低,证实了强金属-载体界面相互作用对局部配位环境的有效调控;
4) 在0.5 M H2SO4电解质中,HSS-CoOx/GDY仅需221 mV和301 mV的过电位即可达到10和100 mA cm-2的电流密度,并可稳定运行超过400小时;将其组装为PEMWE膜电极后,仅需1.80 V即可实现1.0 A cm-2的电流密度,并在200 mA cm-2下稳定运行100小时,衰减速率仅为纯Co3O4的2.65%;
5) 原位电化学阻抗谱和脉冲伏安法揭示了高自旋态调控同步提升催化活性与稳定性的内在机制,HSS-CoOx/GDY中高自旋Co3+诱导的平行自旋排列显著加速了电荷转移,电荷寿命大幅缩短,从而抑制了电荷积累并避免了Co3+→Co4+的过度氧化过程。
Xinyu Ping, et al., High-spin transition metal atoms drive acidic oxygen evolution reactions, Nat. Commun., doi: 10.1038/s41467-026-69682-9.

识别二维码访问原文
5. Journal of the American Chemical Society:双描述符指导稳定酸性析氧金属掺杂RuO2催化剂的智能筛选
为提升RuO2的酸性OER稳定性,研究者开发了多金属氧化物、异质元素掺杂、形貌调控和应变工程等多种策略。然而,目前掺杂剂的选择主要依赖经验试错,缺乏统一的理论描述符来高效筛选稳定组分。密度泛函理论计算虽能用于评估形成能或脱金属能,但这些模型常忽略氧覆盖与氧贫乏环境等表面氧化学计量对掺杂构型的影响。而现有的Pourbaix图分析多局限于单一催化剂体系的垂直案例研究,尚未扩展到元素依赖的跨体系比较以提取定量描述符。此外,Ru-O键长作为结构稳定性指标仅在静态清洁表面上计算,未能捕捉OER过程中动态中间体对键长的演变影响。因此,开发普适且准确的理论描述符,系统评估掺杂对RuO2稳定性的影响,成为该领域亟待解决的关键科学难题。
鉴于此,北京化工大学孙晓明教授、李亚平教授和徐海军教授等人通过第一性原理计算,系统构建了多种金属掺杂RuO2(M-RuO2)在三种氧配位环境(富氧、化学计量、缺氧)下的表面形成能,提出了以稳定性区域(SR)和Ru*O−O/RuOv−O键长为双描述符的筛选框架,成功识别出Rh、Zn、Ga、Bi、Mn、Nb、Sn、Os等8种最优掺杂元素,并通过实验验证了该筛选策略的可靠性。这项工作通过结合第一性原理Pourbaix分析和Ru-O键长调节,揭示了M-RuO2稳定性的热力学和结构起源,为耐用OER电催化剂的理性设计提供了可推广的策略。
本文要点:
1) 首先将M-RuO2表面划分为氧覆盖(M-RuO2-Oc)、化学计量(M-RuO2)和氧亏损(M-RuO2-Od)三种构型,通过形成能比较揭示了不同价态金属的热力学偏好:V、Cr、Nb、Mo、W等高价过渡金属倾向稳定氧覆盖表面,Co、Ni、Zn等低价金属则倾向形成氧亏损结构;
2) 首次构建了跨元素M-RuO2的计算Pourbaix图,通过对不同pH条件下RuO2氧化为RuO4的电位与还原为低价Ru物种的电位之差进行积分定义了SR作为热力学描述符,SR与掺杂元素的三重点pH及相变电位密切相关,其中RuO2氧化为Ru4的电位(E0)越高、还原为Ru3+的电位(E0′)越低、三重点pH(pHtp)越小,则SR值越大,代表催化剂在酸性OER条件下的热力学稳定性越强;
3) 确定了Ru*O-O(Ru位点吸附氧中间体)和RuOv-O(Ru位点相邻氧空位)键长作为结构描述符,其中Ru*O-O键长>1.97 Å时有利于抑制RuO4过度氧化,RuOv-O键长<1.99 Å时则有利于抑制Ru3+溶出,二者共同构成了结构稳定窗口;
4) 将SR与Ru-O键长绘制火山图,筛选出位于火山顶峰的8种最优掺杂元素(Rh、Zn、Ga、Bi、Mn、Nb、Sn、Os),它们同时满足热力学稳定和结构稳定的双重要求,后续实验证实Zn-和Ga-RuO2在100 mA cm-2下可稳定运行650和730小时,衰减速率低至0.37和0.33 mV h-1;
5) 最后通过溶胶-凝胶法合成了Ti、V、Cr、Fe、Ni、Cu、Zn、Ga、Mo、W掺杂的RuO2纳米颗粒,并通过X射线衍射图样、X射线光电子能谱验证了掺杂构型与理论预测的一致性,通过电化学测试验证实验衰减速率与计算SR值的线性关系,证实了双描述符框架的普适性和预测准确性。
Aiqing Cao, et al., Dual-descriptor-guided screening of stable metal-doped RuO2 catalysts for acidic oxygen evolution, J. Am. Chem. Soc., doi:10.1021/jacs.5c17145.

识别二维码访问原文
6. Angewandte Chemie:Re、Ru共掺杂过渡金属合金双功能催化剂实现甘油电氧化合成甲酸酯耦合制氢
甘油电氧化(GOR)作为利用可再生电能从生物质原料合成高值化学品的创新策略,被认为是替代OER的一种极具前景的阳极反应。目前,GOR面临高工作电位、有限的产物选择性和法拉第效率(FE)等问题,严重阻碍了其工业化应用。相比贵金属基催化剂,Ni基催化剂因资源丰富、价格低廉和耐腐蚀性而被广泛研究,然而其活性受限于活性位点Ni3+的高生成电位。同时,竞争性OER以及甲酸盐的进一步氧化导致FE和选择性偏低。过渡金属合金通过各元素间的协同效应可有效提升Ni基催化剂的活性,但难以抑制OER和甘油的过度氧化。对此,杂原子掺杂可调控局部电子结构并产生更多催化活性位点,而有望解决这一问题。Ru物种能促进M3+-OOH活性物种的快速生成,但其在高电位下仍难以避免OER竞争和产物过度氧化。Re物种有望抑制OER和甘油过度氧化,但Re与Ru之间的配位相互作用及其对电子结构的调控仍有待深入揭示。
鉴于此,内蒙古大学武利民教授、王蕾教授和高瑞廷教授等人首次构建了Re和Ru共掺杂的过渡金属合金催化剂(NiCoFeRuRe),用于高效电氧化甘油制甲酸盐。得益于Ru诱导的 M3+-OOH快速生成以及Re物种对OER和甘油过度氧化的有效抑制,该双功能催化剂在甘油氧化耦合产氢体系中展现出优异的催化活性和稳定性。这项工作为理性设计高性能的GOR催化剂提供了一种简便方法。
本文要点:
1) 采用一步电沉积法成功制备了面心立方合金相NiCoFeRuRe层状纳米片催化剂,Ni、Co、Fe、Ru、Re元素均匀分布,证实了Ru和Re的成功共掺杂;
2) X射线光电子能谱和X射线吸收谱结果表明,Ru和Re主要以合金态存在,且Re优先与Ru配位,有效调控了Ru和NiCoFe的电子结构,Ru和Re的共掺杂诱导了Ni3+和Co3+
3) 在1 M KOH + 0.1 M甘油电解液中,NiCoFeRuRe达到10 mA cm-2所需电位仅1.133 V,甲酸盐法拉第效率高达95.64%、选择性接近100%,产率达1.36 mmol cm-2 h-1,并可稳定运行超过450小时;
4) 在1 M KOH中,NiCoFeRuRe的HER过电位仅67 mV(10 mA cm-2),并可稳定运行350小时,将其组装为两电极电解槽用于GOR耦合HER,仅需1.304 V即可达到10 mA cm-2,比传统水电解低293 mV,甲酸盐法拉第效率达95.51%,产氢法拉第效率接近100%,并可稳定运行350小时,产物收益为原料成本的4.99倍;
5) 原位拉曼光谱、原位红外吸收光谱和差分电化学质谱结果表明,Ru的引入使M3+-OOH活性物种在更低电位下生成,而Re的掺杂有效抑制了高电位下CO2和O2的生成,证实Re同时抑制了OER和甲酸盐的过度氧化;
6) 密度泛函理论计算结果表明,Ru和Re的共掺杂使甘油脱氢决速步能垒大幅降低,促进了C-C键断裂,并降低了NiCoFe在费米能级处的电子密度,削弱了金属与中间产物的结合能,从而协同提升了GOR和HER催化活性。
Hengyi Chen, et al., Re and Ru co-doped transition metal alloy as a bifunctional catalyst for electrooxidation of glycerol to formate coupled with H2 production, Angew. Chem. Int. Ed., doi:10.1002/anie.202501766.

识别二维码访问原文
7. Angewandte Chemie:原位X射线发射光谱揭示乙二醇电氧化中晶场介导的钴自旋态演化
钴基羟基氧化物(CoOOH)因其优异的电化学性能,成为EGOR的有力候选催化剂。然而,由于目前研究对反应机理的理解不足,尤其是活性中心电子结构的动态演化规律尚不明确,仍无法解决CoOOH在EGOR中的产物选择性低问题。一般而言,调控第一行过渡金属的3d电子构型,特别是自旋态,有望优化含氧中间体的吸附与转化,然而目前的配位环境调整、异质结构工程等自旋态调控策略仍难以在工况下实时捕获自旋态演化过程。此外,杂原子诱导的晶格畸变与电催化性能之间的关联尚未得到系统研究,而晶格畸变可在晶面间产生拉伸或压缩应变,从而在同一体系中实现双功能反应位点,相关研究尚不充分。当d轨道电子重新分布并跃迁至eg轨道时,电子云的空间取向从面向配体转变为沿坐标轴指向配体,这种空间重排使eg轨道能与吸附的乙二醇中间体的C-C键实现强而有效的轨道重叠。然而,目前自旋态极化对电催化行为影响的深层机制仍不清楚。
鉴于此,安徽大学李鹏教授和柳守杰教授等提出了一种“预催化剂原位重构”策略,通过磷掺杂诱导CoOOH晶格畸变与电子重构,成功构建了高自旋Co3+活性相,并利用原位X射线发射光谱首次动态捕获了钴自旋态从低自旋向高自旋的演变过程,揭示了高自旋Co3+的eg*轨道非简并特性对C-C键选择性断裂的促进作用。这项工作为控制多元醇电氧化路径提供了一种有效的策略。
本文要点:
1) 采用磷掺杂钴钼酸盐预催化剂(P-CoMoO4-x),在碱性电解质中通过原位电化学重构成功制备了P-CoOOH/NF催化剂,其保持纳米棒形貌并表面形成多孔纳米片,磷掺杂诱导了显著的晶格畸变和表面粗糙化,增大了电化学活性面积;
2) 原位X射线发射光谱、X射线吸收光谱和磁性测试结果表明,磷掺杂成功诱导CoOOH发生晶格畸变与电子重构,驱动Co中心自旋态从低自旋向高自旋转变,高自旋Co3+具有非简并的eg*轨道和增大的3d能带分裂,有利于与乙二醇中间体实现最佳轨道重叠,而P-CoOOH中高自旋Co3+占比增加,共同促进了C-C键的选择性断裂;
3) 在1.0 M KOH + 1.0 M EG电解质中,P-CoOOH/NF在10 mA cm-2电流密度下的EGOR电位仅1.26 V,比OER过电位低230 mV,甲酸盐法拉第效率高达93.7%,并可稳定运行160小时以上,性能优于未掺杂CoOOH和多数非贵金属EGOR催化剂;
4) 原位红外光谱和密度泛函理论计算结果表明,反应过程中C-C键逐步断裂,磷掺杂将决速步能垒降低,高自旋Co3+的d带中心上移,费米能级附近电子态密度增大,进而优化了反应物吸附与电子转移;
5) 将P-CoOOH/NF应用于实际PET废碱解液,可在1.41 V下电解20小时实现94.37%的PET升级回收效率,产出105 mM EG转化、45.3 mM甲酸盐和5.2 g高纯对苯二甲酸,所构建的太阳能驱动EGOR-HER耦合系统的产氢法拉第效率可达98.9%。
Xinyue Xu, et al., Unraveling the crystal-field-mediated cobalt spin-state evolution for electrocatalytic ethylene glycol oxidation by in situ x-ray emission spectroscopy, Angew. Chem. Int. Ed., doi:10.1002/anie.4512481.

识别二维码访问原文
8. Advanced Functional Materials:Ce-Mo双调控亚稳态Ni2P相变实现高效尿素电解
在反应中原位生成高活性NiOOH物种的镍基催化剂是最有前景的UOR候选材料之一,然而其实际应用仍受到多种关键问题的制约。首先,镍基催化剂对尿素完全矿化的选择性相较于竞争性OER路径偏低,然而实现工业所需的高电流密度(>100 mA cm-2)往往需要高的过电位,导致能量效率大幅下降。此外,镍基催化剂在长期阳极极化下由于自身不可控的相转变动力学和低效的电子-质子耦合转移过程易发生结构退化。为解决上述问题,掺杂工程、界面设计和非晶化等策略层出不穷,但这些方法往往难以兼顾活性和稳定性。更重要的是,即使是目前最先进的掺杂Ni基催化剂,仍受困于Ni2+向Ni3+转变的预氧化过程。在此背景下,镍磷化物,特别是具有混合价态Ni2+位点的Ni2P,展现出活性、稳定型两全的巨大潜力,但传统磷化方法难以兼顾相纯度与结构精细控制,导致热力学稳定的Ni3P相或块状材料。因此,亟需开发可整合动力学选择性与热力学稳定性的新型磷化路径,以实现Ni2P的原子级精确合成。
鉴于此,哈尔滨工业大学于永生教授、李蒙刚教授和吉林师范大学李嘉明博士等通过Ce和Mo双调控策略,利用Ce3+/Ce4+氧化还原循环触发氧空位形成并引导Ni前驱体向亚稳态Ni2P转变,同时Mo6+作为电子受体优化Ni位点的电子结构,成功构建了高性能Ce/Ni2-xMoxP催化剂用于高效尿素电解。这项工作从原子尺度上阐明了Ce-Mo协同调控电子结构和相变的机理,为设计高效的非贵金属电催化剂和推进尿素电解工业化提供了新的策略。
本文要点:
1) 采用水热-磷化两步法制备了Ce/Ni2-xMoxP催化剂,Ce掺杂诱导Ni基体从四方相Ni3P向六方相Ni2P的结构转变,且当Ce达到0.1 mmol时实现纯六方Ni2P相,各元素均匀分布;
2) X射线光电子能谱和理论结果表明,Ce以Ce3+/Ce4+混合价态存在并促进氧空位生成,Mo以Mo6+形式充当电子受体从Ni的d轨道吸取电子,Ce/Ni2-xMoxP在费米能级处的态密度显著高于Ni3-xMoxP,Ce-Mo双调控增强了Ni与P之间的电子耦合,从而稳定了Ni2P相并提升了电子导电性;
3) 在1 M KOH + 0.33 M尿素电解液中,Ce/Ni2-xMoxP达到100和500 mA cm-2电流密度所需的电位仅1.32 V和1.45 V,远低于Ni3-xMoxP和Ni3P,并可稳定运行超过200小时且活性几乎不衰减;将其作为双功能催化剂组装的尿素电解槽,仅需1.56 V和1.68 V即可达到100和400 mA cm-2的电流密度,并可在500 mA cm-2下稳定运行超过460小时,衰减速率仅0.11 mV h-1;
4) 原位拉曼光谱和原位红外光谱结果表明,Mo的强氧亲和力促进Ni-O键的形成,降低了Ni3P的相转变电位,而Ce的引入进一步通过Ce3+/Ce4+氧化还原诱导晶格重构,降低Ni2P的相转变电位,Ce-Mo双调控增强了界面水分子与活化Mo位点的相互作用,并有效促进了UOR过程中的脱氢和脱羧过程,最终实现了优异的UOR活性;
5) 密度泛函理论计算结果表明,Mo的强吸电子效应和Ce的电子缓冲作用协同增大了Ni位点的空d轨道密度,使Ni的d带中心上移并优化了对尿素分子的吸附能力,同时Mo的高氧亲和力加速了质子-电子耦合步骤,Ce的氧化还原循环为反应中间体提供了额外的电子缓冲通道,从而显著降低了UOR的理论过电位。
Longyu Qiu, et al., Metastable Ni2P phase transformation via Ce-Mo dual-modulation for efficient urea electrolysis, Adv. Funct. Mater., doi:10.1002/adfm.75427.

识别二维码访问原文
顶刊速报|中国科学院理化所吴骊珠院士、中科大俞书宏院士等电解水催化剂最新成果速览
顶刊速报|上海交通大学宋钫教授、南开大学焦丽芳教授、圣路易斯华盛顿大学武刚教授等电解水催化剂最新成果速览
顶刊速报|美国康奈尔大学Héctor D. Abruña院士、长春应化所邢巍研究员、哈工大王振波教授等氧还原电催化最新成果速览
顶刊速报|广西大学刘熙俊教授、温州大学金辉乐教授、北京大学郭少军教授等贵金属电催化剂最新成果速览

随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- #深圳坪山上门做饭
- 当一个女人来到深圳,就会变成一个更强大的女人
- 广东国际学校|深圳市承翰学校(国际课程实验校区),Cognia+AP+A-Level 三认证,全球认可
- 深圳翻身公益相亲|大厂精英专属脱单平台 专为华为、腾讯、阿里、微软、科通优质单身打造纯公益初心,全程无红娘中介费、无隐形消费
- 覆盖全技术方向!深圳优质 IT 岗位整理,12K 起步福利齐全!!!
- 深圳湾口岸 精致4房出租 随时入住
- 深圳蛇口海上世界明华轮
- 今日有點無聊想過來深圳走走[疑問]
- 深圳大鹏|单身交友轰趴一日游|逃离城市喧嚣,遇见同频的人
- 在深圳,居然藏着一座1700岁的古城,是周末休闲游玩的好去处
