深圳大学Angew Chem:揭示价态驱动的电荷补偿机制以指导钌基催化剂在酸性水电解中的相工程
- 2026-05-16 08:30:09
核心机制:提出价态驱动的电荷补偿机制,首次实现 Ru 基催化剂相结构的确定性调控。
三类掺杂 → 三种相结构
四价金属(Hf、Mn、Sn):电荷匹配 → 单相 RuO₂
三价金属(Cr、Fe、Ga):电荷失配 → RuO₂ + Ru 混合相(最优)
二价金属(Ni、Cu、Zn):严重失配 → RuO₂ + MO 相分离
最佳催化剂:RuGa
酸性 OER 过电位仅 180 mV @ 10 mA·cm⁻²
稳定性 >500 小时(无明显衰减)
在 PEMWE 中:1.63 V 达到 1 A·cm⁻²
100 小时 稳定运行 @ 500 mA·cm⁻²
理论 + 实验双重验证
DFT:形成能、掺杂能、Bader 电荷、氧空位形成能、相分离自由能
HAADF-STEM、XRD、XPS、XANES/EXAFS 全面证实相结构与电荷补偿路径
设计启示:三价掺杂可诱导金属 Ru 析出,构建 Ru/RuO₂ 界面,是打破活性-稳定性权衡的有效策略。
2 价态驱动的相调控机制
本文提出一种价态驱动的电荷补偿机制,通过调控掺杂金属的价态实现对 Ru 基催化剂相结构的精准控制。如图 Figure 1 所示,当采用四价金属(Hf、Mn、Sn)掺杂时,因 M⁴⁺ 与 Ru⁴⁺ 价态相同且氧化物均为金红石结构,晶格失配度仅约 5%(Figure 2a),八面体畸变也小于 5%(Figure 2b),因此无需电荷补偿即可形成单相 RuO₂ 固溶体。DFT 计算表明,四价掺杂的形成能(−3.29~−3.42 eV)和掺杂能(0.55~1.83 eV)均为最低(Figure 2c, d),且氧空位形成能很高(3.55~4.94 eV,Figure 2f),说明体系几乎没有产生氧空位的倾向。Bader 电荷分析(Figure 2e)显示 Ru 的电子损失约为 1.67–1.68 e⁻,Ru 保持 +4 氧化态。
当采用三价金属(Cr、Fe、Ga)掺杂时,M³⁺ 取代 Ru⁴⁺ 会引入净负电荷。体系首先通过提高邻近 Ru 的氧化态(Bader 电荷显示 Ru 电子损失增至 1.68–1.70 e⁻)以及生成氧空位(氧空位形成能降至 2.61–3.08 eV,Figure 2f)进行初步补偿。然而在高掺杂浓度下,这两种途径不足以完全平衡电荷,系统被迫启动第三种补偿路径:Ru⁴⁺ 被还原为 Ru⁰,并析出为独立的金属 Ru 相。吉布斯自由能计算(Figure 2h)证实,对于三价掺杂,Ru⁴⁺→Ru⁰ 的自由能变化(−1.25~−2.73 eV)远低于 M⁰→M₂O₃ 的 ΔG(−0.08~−0.50 eV),因此最终形成 RuO₂–Ru 混合相。这一结果也与结构分析一致:虽然三价金属氧化物(Cr₂O₃、Fe₂O₃、Ga₂O₃)与 RuO₂ 的晶格失配度 >30%(Figure 2a),但其八面体单元畸变仍 ≤5%(Figure 2b),表明掺杂原子优先进入 RuO₂ 晶格而非形成独立氧化物相。
对于二价金属(Ni、Cu、Zn)掺杂,每个 M²⁺ 取代 Ru⁴⁺ 带来两倍于三价掺杂的电荷失配。体系即使大量产生氧空位(氧空位形成能低至 1.67–2.07 eV,Figure 2f)并大幅提高 Ru 氧化态(Ru 电子损失达 1.72–1.75 e⁻,Figure 2e),仍无法维持晶格稳定。自由能计算(Figure 2g)表明,M⁰→MO 的 ΔG(−4.82~−7.41 eV)远低于 Ru⁴⁺→Ru⁰ 的 ΔG(−0.14~−0.99 eV),因此体系倾向于发生相分离,形成 RuO₂–MO 混合相(MO 如 NiO、CuO、ZnO)。这与二价金属氧化物与 RuO₂ 之间约 12% 的晶格失配(Figure 2a)和略高的八面体畸变(Figure 2b)相一致。
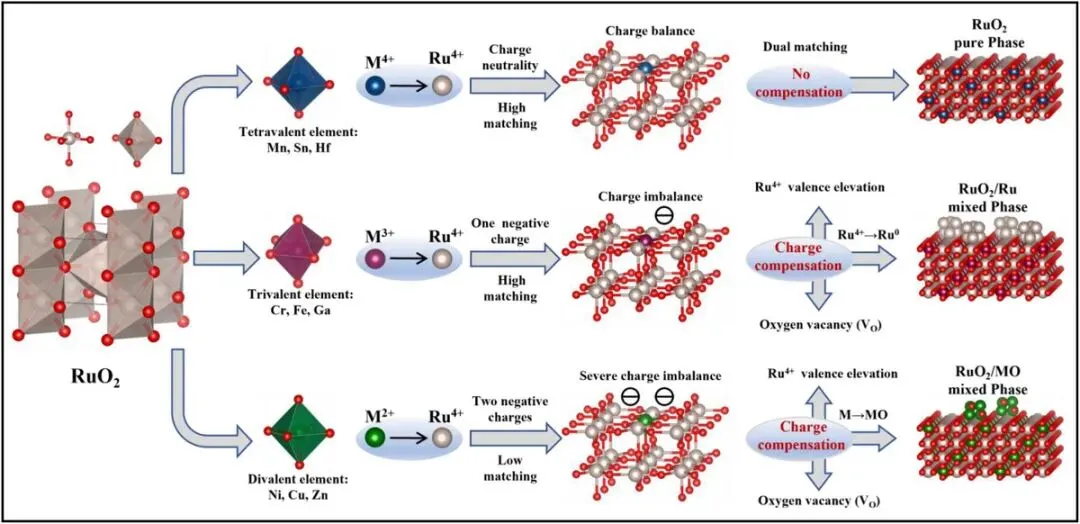
图1
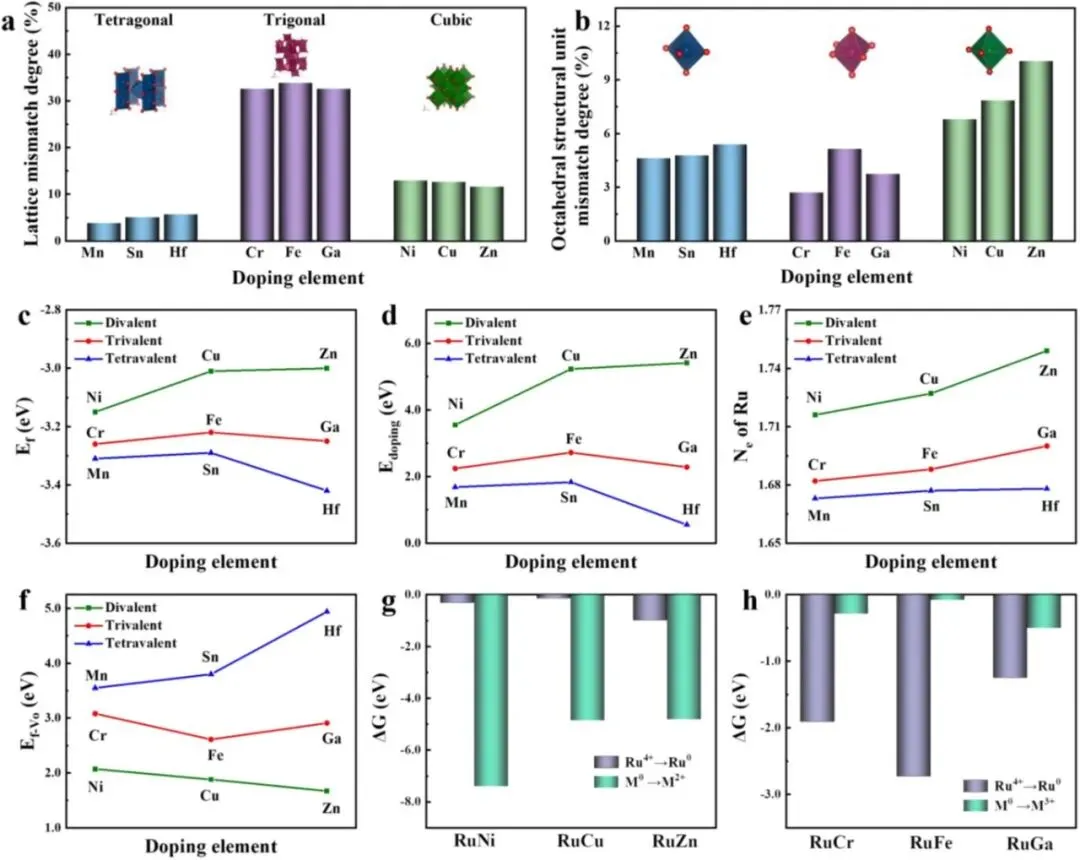
图2
3 DFT 计算对价态驱动机制的理论验证
为了从理论上验证上述价态驱动的相调控机制,本文针对一系列 RuM 催化剂(M = Mn, Sn, Hf, Cr, Fe, Ga, Ni, Cu, Zn)开展了系统的 DFT 计算,重点考察了催化剂的形成难易程度及其对应的电荷补偿路径。
首先,形成能(Figure 2c)和掺杂能(Figure 2d)的计算结果清晰支持了所提出的机制。四价掺杂金属(Mn, Sn, Hf)表现出最低的形成能(分别为 –3.31、–3.29 和 –3.42 eV)和最低的掺杂能(分别为 1.68、1.83 和 0.55 eV),表明它们最易于掺入 RuO₂ 晶格,并倾向于形成单相固溶体。与之相反,二价掺杂金属(Ni, Cu, Zn)具有最高的形成能(–3.15、–3.01 和 –3.00 eV)和最高的掺杂能(3.55、5.23 和 5.41 eV),证实其掺杂难度最大。三价掺杂金属(Cr, Fe, Ga)的各项能量值则处于中间位置(形成能 –3.26、–3.22、–3.25 eV;掺杂能 2.24、2.72、2.28 eV),与其适中的掺杂可行性相一致。
随后,Bader 电荷分析(Figure 2e)进一步揭示了掺杂原子与 Ru 之间的电子转移情况,直接反映了 Ru 氧化态的调控幅度。对于四价掺杂(Mn, Sn, Hf),Ru 的电子损失数(Nₑ)分别为 1.673、1.677 和 1.678 e⁻,变化较小。三价掺杂(Cr, Fe, Ga)使 Ru 的电子损失略有增加,分别达到 1.682、1.688 和 1.700 e⁻,表明 Ru 的氧化态轻微升高以补偿电荷失衡。而二价掺杂(Ni, Cu, Zn)则导致 Ru 的电子损失显著增至 1.716、1.727 和 1.749 e⁻,说明严重的电荷失配大幅提升了 Ru 的氧化态。这一趋势明确显示:掺杂金属价态越低,Ru 的氧化态升高越显著。
接下来,氧空位形成能(E_fᵥₒ) 的计算结果(Figure 2f)为氧空位介导的电荷补偿路径提供了有力证据。四价掺杂的 E_fᵥₒ 值很高(Mn 3.55 eV,Sn 3.80 eV,Hf 4.94 eV),表明氧空位难以形成,这与四价掺杂几乎不需要电荷补偿的结论一致。三价掺杂的 E_fᵥₒ 值明显降低(Cr 3.08 eV,Fe 2.61 eV,Ga 2.61 eV),有利于通过生成氧空位进行二次补偿。二价掺杂则呈现出最低的氧空位形成能(Ni 2.07 eV,Cu 1.88 eV,Zn 1.67 eV),证实其极强的氧空位形成倾向。
值得强调的是,上述两种补偿途径(提高 Ru 价态和生成氧空位)仅在中等掺杂浓度下有效。在高掺杂浓度下,电荷失衡会显著加剧。如果仅依赖前两种途径,体系能量将持续升高,导致结构失稳。为了降低体系能量,系统会自发启动第三种电荷补偿途径——相分离。
为了厘清三价和 二价掺杂的具体相分离路径,本文计算了两个可能过程的吉布斯自由能(ΔG)(Figure 2g, h)。对于二价金属掺杂(Ni, Cu, Zn),M⁰ → MO 反应的 ΔG 值(–7.41、–4.86、–4.82 eV)远低于 Ru⁴⁺ → Ru⁰ 的 ΔG(–0.31、–0.14、–0.99 eV),证明生成 MO 相(如 NiO、CuO、ZnO)是热力学上有利的分离方式。而对于三价金属掺杂(Cr, Fe, Ga),Ru⁴⁺ → Ru⁰ 的 ΔG 值(–1.91、–2.73、–1.25 eV)远低于 M⁰ → M₂O₃ 的 ΔG(–0.29、–0.08、–0.50 eV),证实析出金属 Ru 相是主导的相分离路径。
4 实验验证:结构表征
为了从实验上验证上述价态驱动的相调控机制,本文选取了三个代表性体系进行详细研究:RuHf(四价掺杂,预期单相)、RuGa(三价掺杂,预期 RuO₂–Ru 混合相) 以及 RuCu(二价掺杂,预期 RuO₂–MO 混合相)。所有 RuM 催化剂均采用溶胶-凝胶法合成,结构表征结果与理论预测高度一致。
首先,高角环形暗场扫描透射电子显微镜(HAADF-STEM) 图像显示:RuHf(Figure 3a)呈现出清晰的晶格条纹,面间距为 0.32 nm 和 0.26 nm,分别对应于金红石 RuO₂ 的 (110) 和 (101) 晶面,未观察到任何第二相,证实其单相结构。能谱(EDS)面分布图(Figure 3d)显示 Hf 均匀分布,表明成功掺入。对于 RuGa(Figure 3b),HAADF-STEM 中同时存在两套晶格条纹:一套匹配 RuO₂ 的 (110)/(101) 面,另一套面间距为 0.205 nm 和 0.235 nm,分别对应于金属 Ru 的 (101) 和 (100) 晶面,直接证明了 RuO₂–Ru 混合相的形成。EDS 面分布(Figure 3e)证实 Ga 的均匀掺杂。对于 RuCu(Figure 3c),除 RuO₂ 的特征条纹外,还观察到面间距为 0.23 nm 的额外条纹,与 CuO 的 (111) 晶面相匹配,表明形成了 RuO₂–CuO 混合相,EDS 面分布(Figure 3f)也显示 Cu 与 Ru 共存。
其次,X 射线衍射(XRD) 图谱进一步佐证了上述相组成。对于四价掺杂体系(Mn、Sn、Hf),XRD 图谱(Figure 3g)中仅出现金红石 RuO₂ 的衍射峰(JCPDS #40‑1290),无任何杂峰,确认了单相 RuO₂ 结构。对于三价掺杂体系(Cr、Fe、Ga),XRD 图谱(Figure 3h)中除 RuO₂ 峰外,还在 38.4°、42.2° 和 44.0° 处出现金属 Ru 的特征峰(JCPDS #06‑0663),证实了 RuO₂–Ru 混合相的存在。对于二价掺杂体系(Ni、Cu、Zn),XRD 图谱(Figure 3i)中 RuO₂ 峰与相应的 MO 相峰(例如 CuO 在 32.5° 和 38.7° 的峰,JCPDS #48‑1548)共存,验证了 RuO₂–MO 相分离。
为了进一步揭示电荷补偿路径的动态演化,本文开展了时间分辨 XRD 表征,观察不同退火时间下样品的相演变。对于四价掺杂(RuHf),在退火过程中衍射峰始终只对应金红石 RuO₂,未出现任何第二相(Figure 4a)。对于三价掺杂(RuGa),退火 1 小时后金属 Ru 的衍射峰几乎不可见,但随着退火时间延长,Ru 峰的强度逐渐增加(Figure 4b),说明初始补偿主要依赖 Ru 氧化态升高和氧空位生成,而长时间退火后 Ru⁴⁺ → Ru⁰ 的相分离路径逐渐主导。对于二价掺杂(RuCu),退火初期 MO 相峰较弱或不存在,随着退火时间延长,MO 峰强度逐渐增强(Figure 4c),表明初始补偿同样依赖 Ru 氧化和氧空位,但后期 M⁰ → MO 的直接氧化成为热力学主导路径。
X 射线光电子能谱(XPS) 提供了有关氧化态和电荷补偿的进一步信息。高分辨 Ru 3p XPS 谱(Figure 4d)显示:RuHf 的特征双峰位于 462.12 eV(Ru⁴⁺ 3p₃/₂)和 484.23 eV(Ru⁴⁺ 3p₁/₂),证实单相催化剂中 Ru 主要为 +4 价。相比之下,RuCu 的 Ru 3p 峰正向移动了 +0.12 eV,表明 Ru 氧化态升高以补偿二价掺杂带来的严重电荷失配。而 RuGa 的 Ru 3p 峰负向移动了 –0.28 eV,与混合相中部分 Ru⁴⁺ 还原为 Ru⁰ 的现象一致。对三价掺杂体系的谱图进行解卷积(Figure S25 虽未展示,但原文提到),定量得到金属 Ru 的含量分别为 Cr 37.7%、Fe 36.8%、Ga 34.8%。O 1s XPS 谱(Figure 4e)可解卷积为四个组分:晶格氧(~529.11 eV)、氧空位(~530.16 eV)、表面羟基(~531.15 eV)和吸附水(~532.28 eV)。定量分析(Figure 4f)表明,RuCu 和 RuGa 的氧空位含量显著更高(分别为 25.3% 和 24.3%),而 RuHf 仅为 19.6%。这一趋势在所有掺杂体系中一致:四价掺杂引入的氧空位最少,二价掺杂最多,直接证实了氧空位作为关键电荷补偿路径,且其浓度随掺杂价态降低而增加。
最后,X 射线吸收近边结构(XANES) 和 扩展 X 射线吸收精细结构(EXAFS) 提供了局部电子结构和配位环境的信息。Ru K 边 XANES 谱(Figure 4g)显示,RuCu、RuGa 和 RuHf 的吸收边位置均介于 Ru 箔(Ru⁰)和 RuO₂(Ru⁴⁺)之间,表明 Ru 的平均氧化态低于 +4。通过线性组合拟合(Figure 4h)得到的平均价态为:RuCu 约 +3.9,RuGa 约 +3.0,RuHf 约 +3.6,与 XPS 结果吻合。傅里叶变换 EXAFS 谱(Figure 4i)显示所有样品均在 ~1.5 Å 处有一个主峰,对应 Ru–O 散射路径。值得注意的是,RuGa 在 ~2.4 Å 处出现了一个额外的峰,对应于金属 Ru 中的 Ru–Ru 配位,进一步证实了 RuO₂–Ru 混合相的形成。
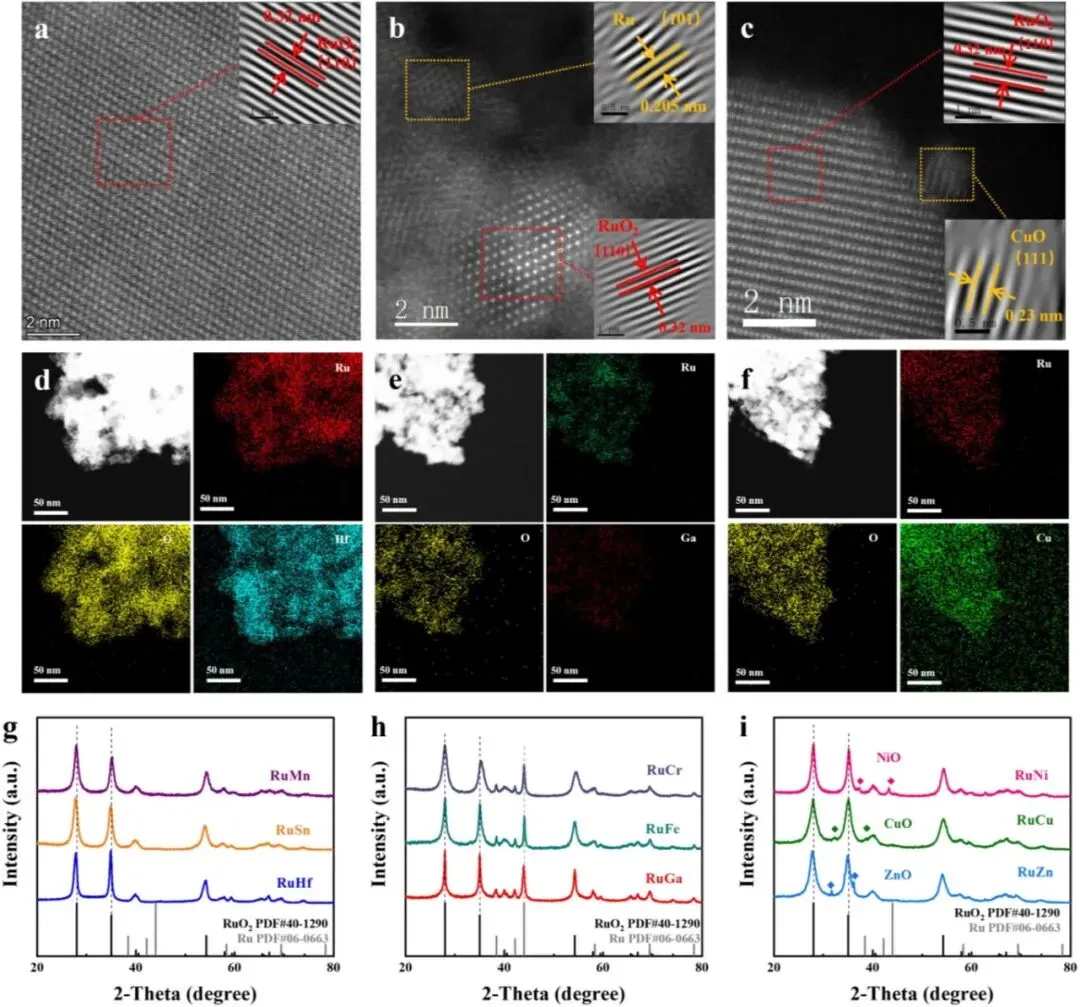
图3
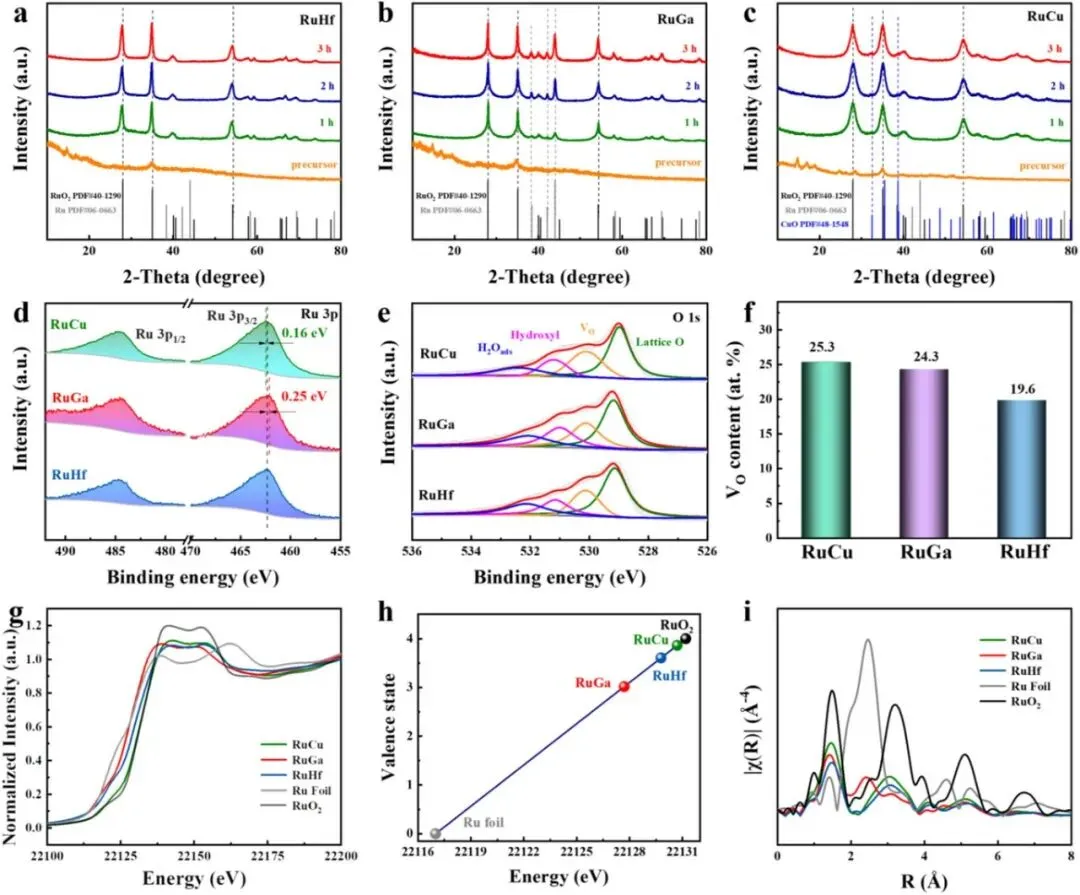
图4
5 RuM 催化剂的电催化性能
本文在 O₂ 饱和的 0.5 M H₂SO₄ 溶液中,采用标准三电极体系评价了所有 RuM 催化剂的酸性析氧反应(OER)性能,并以商业 RuO₂(c‑RuO₂)作为基准。线性扫描伏安(LSV)曲线(Figure 5a–c)显示,所有 RuM 催化剂的性能均优于 c‑RuO₂,后者达到 10 mA·cm⁻² 的电流密度需要 323 mV 的过电位。具体而言,四价掺杂催化剂(Mn、Sn、Hf) 达到 10 mA·cm⁻² 的过电位分别为 194、178 和 225 mV;三价掺杂催化剂(Cr、Fe、Ga) 分别为 198、176 和 180 mV;二价掺杂催化剂(Ni、Cu、Zn) 分别为 177、188 和 198 mV。此外,所有 RuM 催化剂的 Tafel 斜率均低于 c‑RuO₂(112.5 mV·dec⁻¹),表明其 OER 动力学显著增强。这些结果共同证明,价态驱动的掺杂策略能够有效提升 Ru 基 OER 催化剂的活性和反应动力学。
RuM 催化剂还展现出更大的电化学活性表面积,其双电层电容(C_dl)范围为 69.6–101.9 mF·cm⁻²,远高于 c‑RuO₂ 的 39.0 mF·cm⁻²。通过 ECSA 归一化的 LSV 曲线显示,三个代表性样品(RuCu、RuGa 和 RuHf)的本征活性均高于 c‑RuO₂,其中 RuGa 表现出最高的本征活性,这归因于 RuO₂–Ru 混合相结构的协同效应。电化学阻抗谱(EIS)测量进一步表明,RuGa 在三个代表性样品中具有最低的电荷转移电阻,与其加速的 OER 动力学一致。
虽然高 OER 活性至关重要,但优异的长期稳定性对于实际水分解应用同样关键。在 10 mA·cm⁻² 恒电流密度下进行的计时电位(CP)测试(Figure 5d)揭示了巨大的耐久性差异。c‑RuO₂ 在 20 小时内迅速降解。相比之下,掺杂显著提高了稳定性:RuHf 和 RuCu 分别稳定运行约 150 和 350 小时。尤为突出的是,RuGa 表现出卓越的耐久性,在连续运行超过 500 小时内未见明显的电位升高。对反应后 RuGa 的系统表征显示,其形貌、晶格结构、RuO₂–Ru 混合相以及化学状态均保持完好,未发生相变或峰位移。
为了评估实际应用潜力,本文以 RuGa 为阳极催化剂、商业 Pt/C 为阴极催化剂组装了质子交换膜水电解池(PEMWE)(Figure 5e)。在 80 °C 下测得的极化曲线(Figure 5f)显示,RuGa 基膜电极组件(MEA)达到 1 A·cm⁻² 的电流密度仅需 1.63 V 的电池电压,比 c‑RuO₂ 基 MEA(1.83 V @ 1 A·cm⁻²)低了 200 mV。与近期报道的高性能 Ir/Ru 基催化剂对比(Figure 5g),RuGa 位列最活跃的 OER 催化剂之一。该 PEMWE 装置生产 1 kg 氢气的成本仅为 0.874 美元,远低于美国 DOE 的 2 美元目标。此外,在 500 mA·cm⁻² 和 80 °C 下进行的 100 小时耐久性测试(Figure 5h)显示,RuGa 基 MEA 的电压没有明显升高,证明了其优异的运行稳定性和强大的实际应用潜力。
为了深入理解相结构对 OER 性能的调控机制,本文利用 DFT 模拟对 RuM 催化剂进行了机理研究。选取 RuCu、RuGa 和 RuHf 作为代表性样品。如 Figure 6a 和 Figure 6b 所示,第三步(*O → *OOH 的转化)被一致确定为电位决定步骤(PDS),对应的过电位分别为 RuHf 0.86 V、RuGa 0.61 V 和 RuCu 0.65 V。与纯 RuO₂ 相的 RuHf 相比,混合相结构的 RuGa 和 RuCu 表现出显著增强的 OER 活性,其中 RuGa 的性能最高。DFT 预测的活性趋势与实验结果高度一致。此外,为了考察同一相结构下不同掺杂离子的影响,本文还对比了 RuFe 和 RuGa 的自由能图。RuFe 的 PDS 同样是 O → OOH,过电位为 0.59 V,低于 RuGa,这与实验观察到的 OER 活性顺序一致。进一步,Figure 6c 绘制了 ΔGOOH 作为 ΔGOH 的函数。对于 RuHf(纯 RuO₂ 相),OOH 和 OH 的吸附自由能遵循经典线性关系 ΔGOOH = ΔGOH + 3.20 eV(黄线)。然而,对于混合相催化剂(包括 RuGa、RuFe 和 RuCu),由于载体与负载物之间的协同效应,这一线性关系被打破。
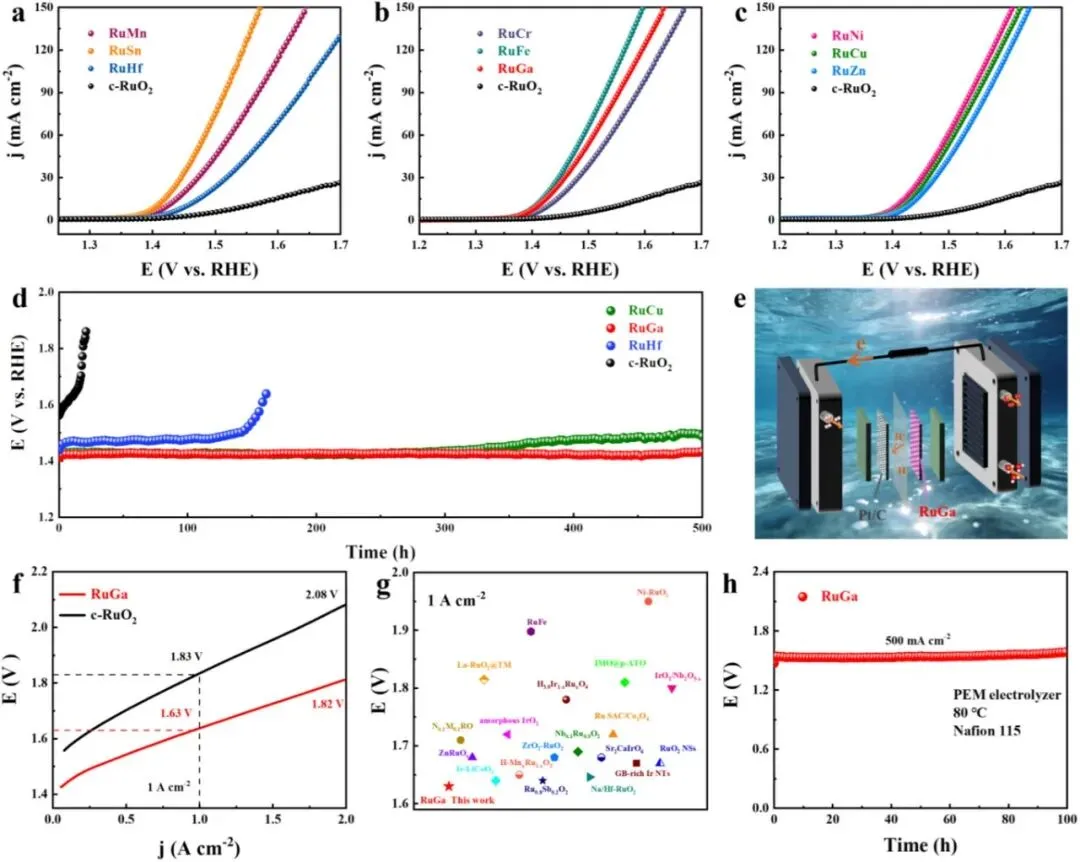
图5
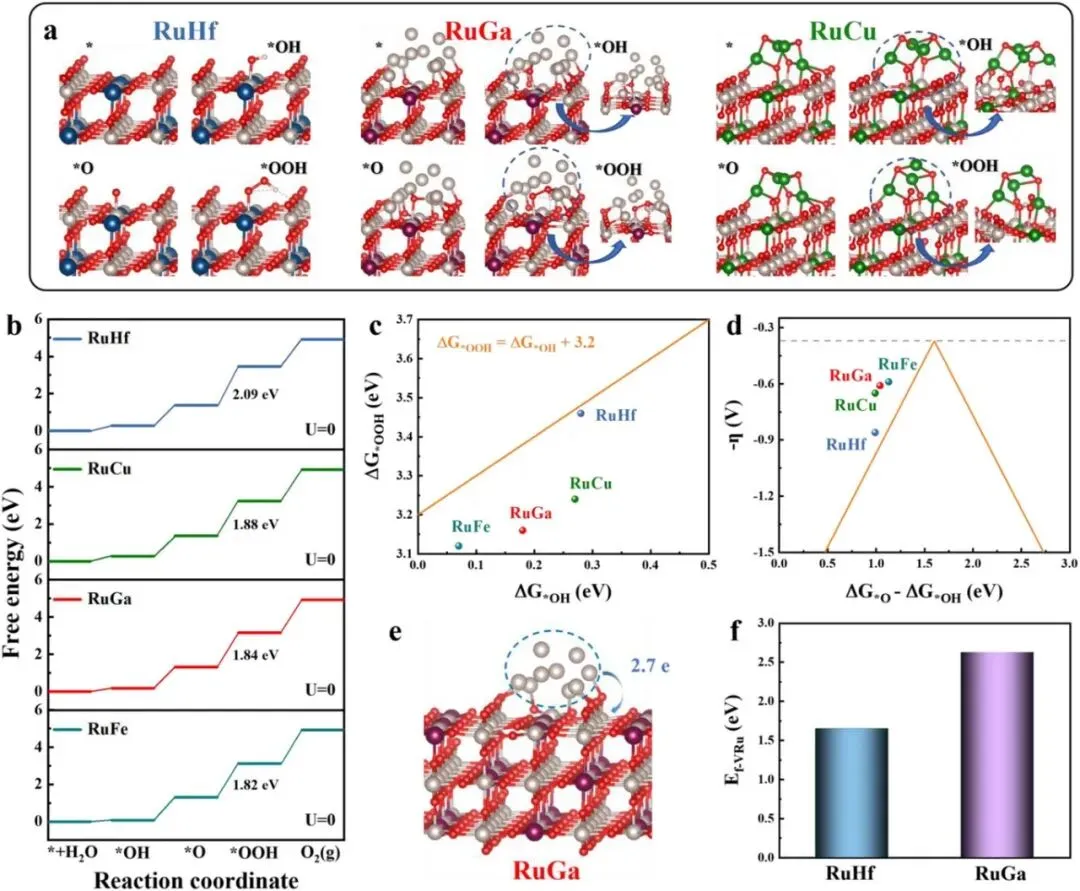
图6
版权声明与须知
- 感谢您关注并阅读本公众号内容。我们始终秉持“分享知识,尊重原创”的理念,致力于为广大科研工作者和学术爱好者提供有价值的前沿解读与学术资讯。
- 本文内容仅为学术分享与知识传播目的,基于已正式发表的学术论文进行中文解读、观点提炼或内容编译。文中所涉及的研究成果、数据图表、核心观点等所有知识产权,均归属于原论文作者及其所在机构,具体详见正文顶部原始引用信息。
- 文中解读仅代表编译者基于个人学术理解所做的转述与分析,不代表原论文作者或出版方的立场。如有解读不准确之处,欢迎学界同仁留言批评指正,我们将虚心学习并及时修正。如内容不慎侵犯了您的合法权益,请第一时间通过后台与我们联系,我们将积极配合处理。
- 我们也诚挚欢迎海内外从事一线研究工作的同行投稿分享您的最新成果或研究亮点,让前沿学术动态被更多人所看见
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 深圳湾设计对话|《世纪线索》展览开幕论坛活动预告
- 【深圳诗词】第816期 | 感悟人生真妙谛,诗香载梦寄毫端
- 【深圳诗词】第815期 | 赖万辉临江仙二十阙
- 【深圳诗词】第803期 | “迎国庆、贺中秋”诗词专辑
- 深圳|北京金诚同达(深圳)律师事务所非诉领域诚聘留用实习生
- 中国检验认证集团深圳有限公司一行到访研究院开展调研交流
- 深圳楼市新政来了 丈夫月薪涨至6万元后频繁家暴“在家五个半月能打四个月” 美国防部正式申请更名为“战争部” “朋友圈改版”冲上热搜
- 【深圳诗词】第811期 | 冬来秋去季如然,顺势而为免问禅
- 【深圳诗词】第812期 | 嘱托殷殷催奋进,老区富路策奔康
- 【深圳诗词】第809期丨重阳荟萃
