江南大学/中科院深圳先进院/华东理工Nat. Commun.:1T′-MoS₂靠面内+边缘位点协同实现CO₂高选择性制甲醇
- 2026-07-20 16:04:24
1、研究背景
CO₂加氢制甲醇是同时解决碳排放和能源存储的重要路线,但这条路不好走。CO₂分子里的C=O键能高达803 kJ/mol,活化本身就需要克服很大的热力学障碍;更麻烦的是,反应中间体*CO的C-O键是断还是留,直接决定产物走甲烷路径还是甲醇路径。也就是说,催化剂既得能打断C=O键活化CO₂,又得能在后续加氢中稳住C-O键不让它断掉,这两个要求其实是矛盾的。
为了解决这个问题,研究人员借鉴自然界酶催化的思路,提出了串联催化—把不同功能的活性位点凑在一起,让中间体在它们之间传递,分步完成反应。比如把Pd/CNT和ZnZrOₓ物理混合,或者Cr/CuO₂配Cu/Mo₂C,概念上能跑通,但实际效果很差。因为粉末混在一起,位点分布完全是随机的,*CO这种中间体刚生成就可能跑丢,根本到不了下一个位点,传输效率成了瓶颈。
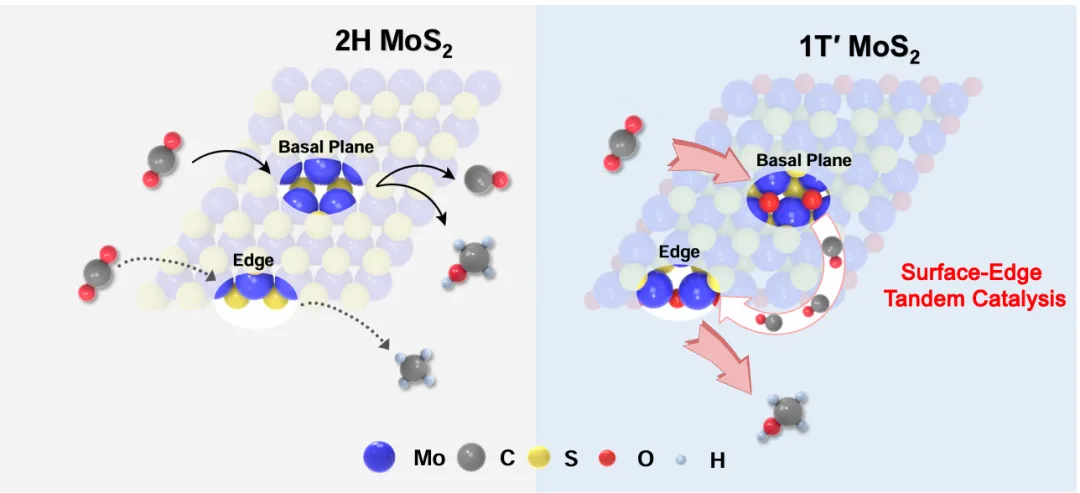
那能不能把两种位点做进同一片材料里,让它们离得近一点?二维过渡金属硫族化合物,特别是MoS₂,天生就适合干这个—它的面内(basal plane)和边缘(edge)是空间分离的,像一张纸的正反面和侧边。但普通2H-MoS₂有个毛病:它的边缘是S边和Mo边混在一起的,活性位点种类杂,很难给不同位点分配不同任务。
1T′-MoS₂就不一样了。它只有S边,没有Mo边,边缘结构单一;而且1T′相里Mo⁴⁺的电子是离域的,材料呈现金属性,d带中心和态密度被独特调制,H₂活化能力比2H相强得多。更关键的是,1T′-MoS₂的二维结构允许你通过氧化处理,在面内造一类缺陷、在边缘造另一类缺陷,让它们各司其职。作者的核心策略就是:利用1T′-MoS₂的相结构和二维形貌,在同一个纳米片上构建"面内解离CO₂、边缘加氢制甲醇"的串联反应路径。
二、研究内容概述
用水热法加葡萄糖做稳定剂合成1T′-MoS₂/C,空气200°C处理3小时得到1T′-MoS₂/C(Air)。表征发现1T′相纯度高达96%,且氧化后形成了面内双氧取代硫缺陷(2OS)和边缘氧化位点(EO-OS)两类活性位点。
三、图表解读
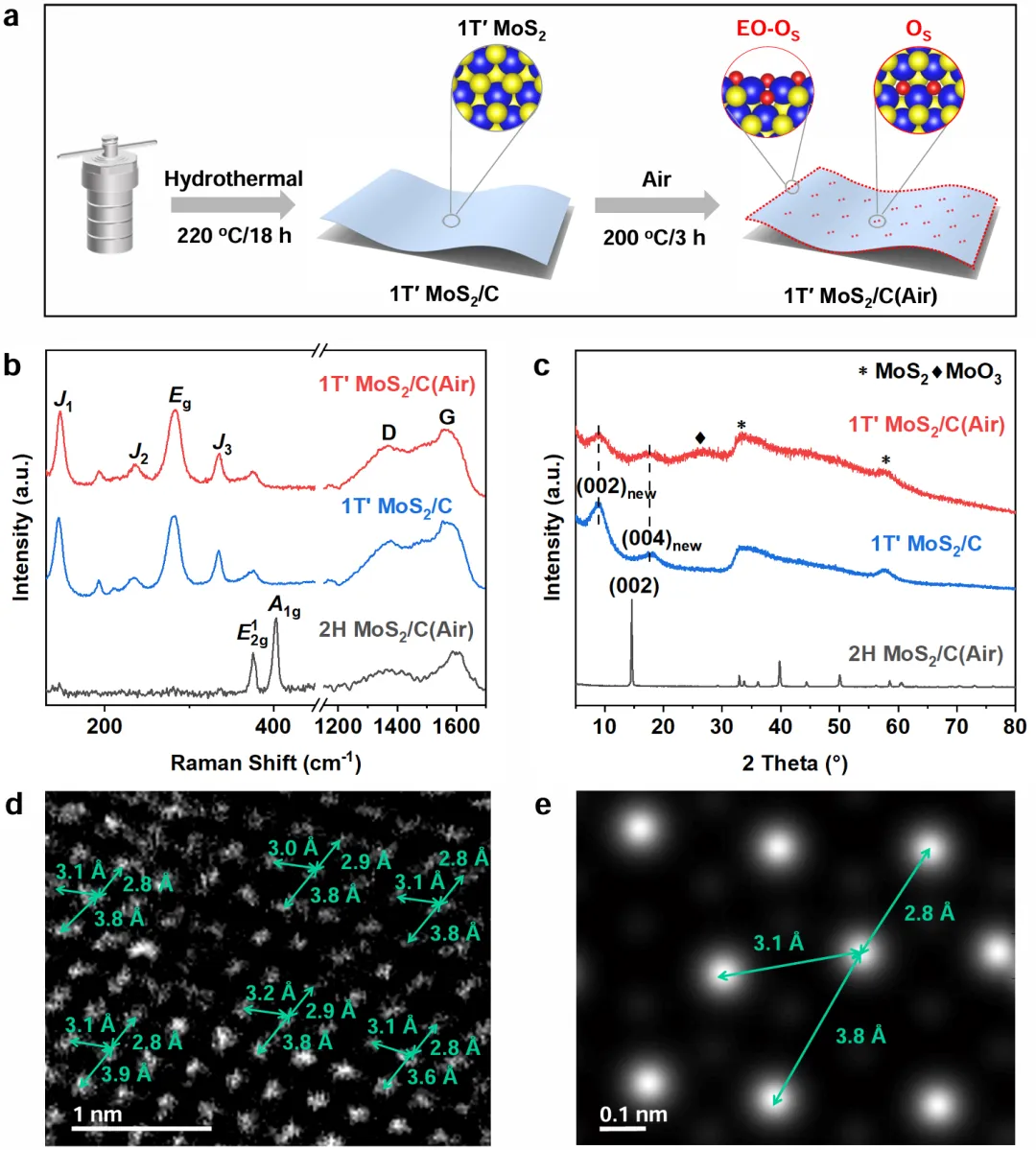
Figure 1是结构确认。Raman在146、234、280、334 cm⁻¹出现1T′相特征峰,XRD在33.5°也有1T′特征峰;球差电镜看到Mo原子排成锯齿链,和1T′相模型一致。
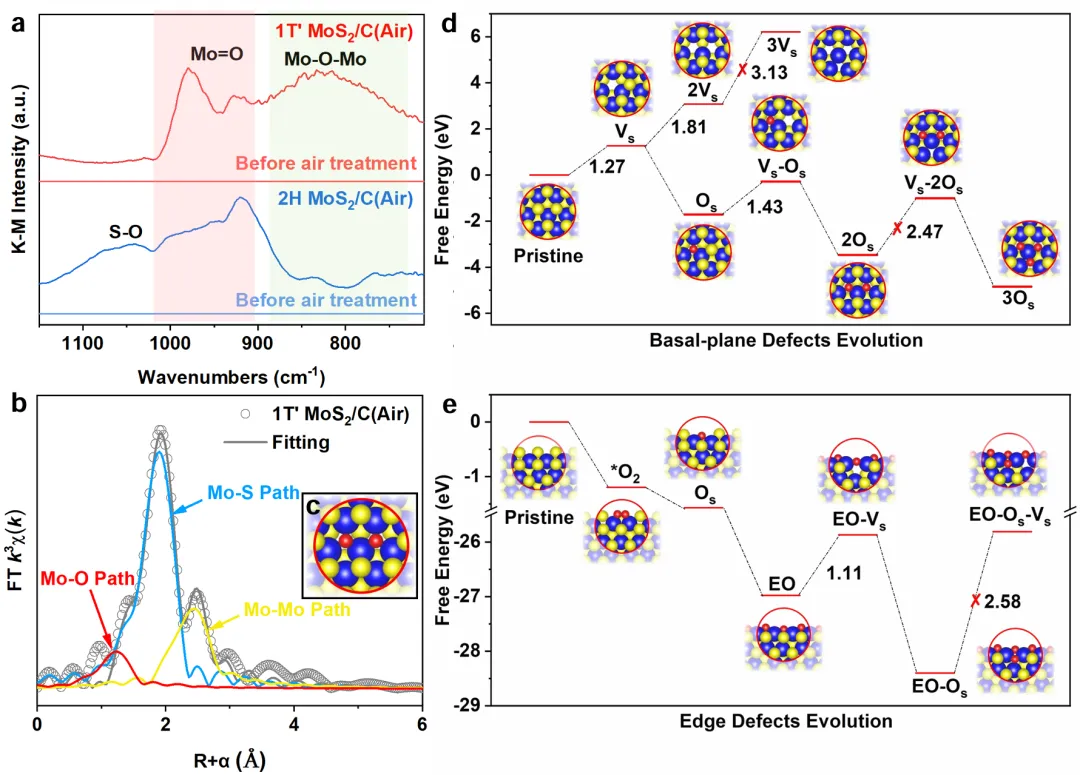
Figure 2讲活性位点怎么来的。原位DRIFTS显示,1T′-MoS₂/C(Air)煅烧后出现边缘Mo=O(900–1000 cm⁻¹)和面内Mo-O-Mo(700–860 cm⁻¹),说明面内和边缘都氧化了;而2H相只有边缘氧化。EXAFS拟合给出五硫一氧配位,结合DFT确认面内主要形成2OS、边缘形成EO-OS。
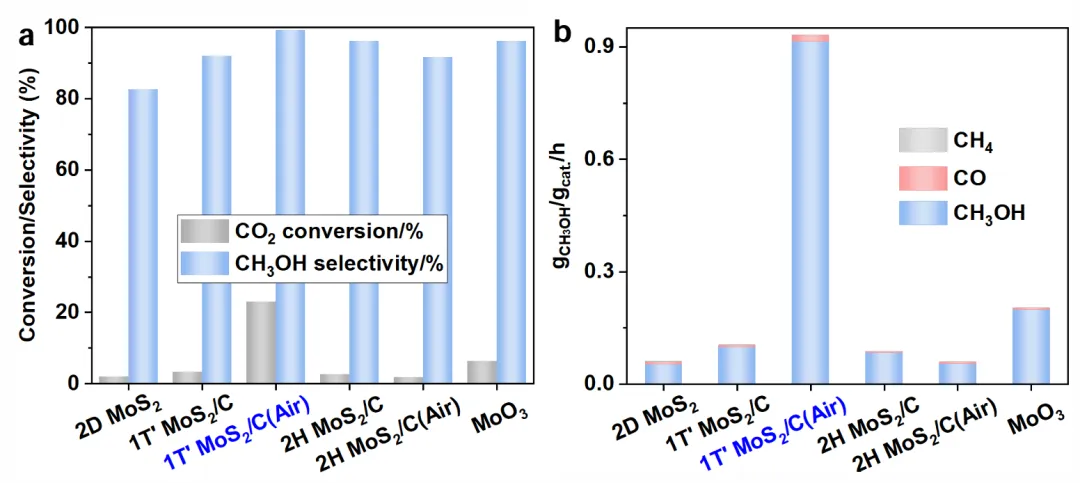
Figure 3看性能。210°C、3.3 MPa、H₂/CO₂=5:1条件下,1T′-MoS₂/C(Air)的CO₂转化率23.0%,甲醇选择性99.2%,比反应速率0.91 g/g/h,远高于2D MoS₂、2H相等对照样。把1T′-MoS₂/C和MoO₃机械混合,选择性仅90.5%,转化率也低近十倍,说明位点必须在空间上紧密耦合。
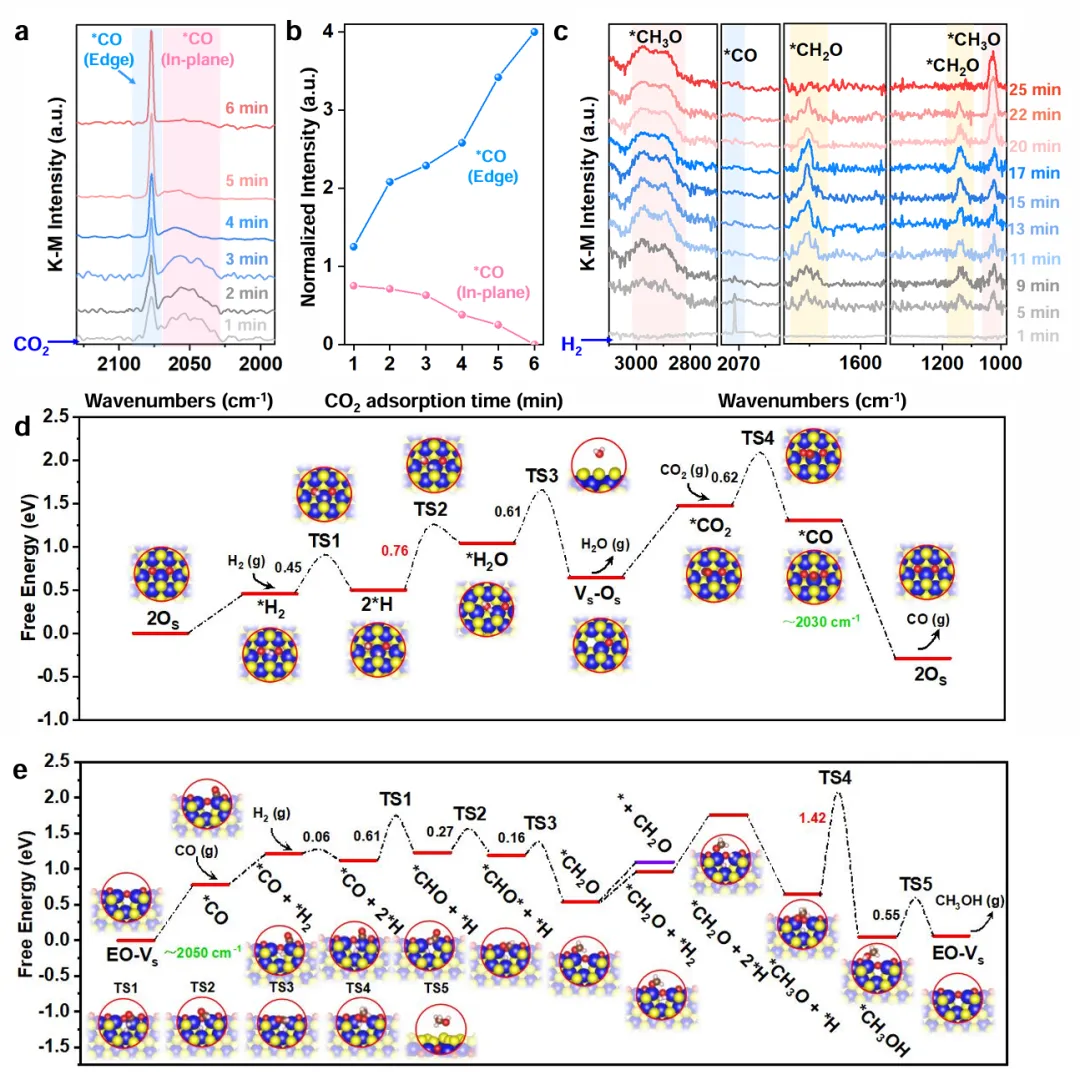
Figure 4揭示串联机理。原位CO₂-DRIFTS显示,面内CO(2054 cm⁻¹)先出现、6分钟后消失,边缘CO(2078 cm⁻¹)逐渐增强,说明CO₂先在面内解离,CO脱附迁移到边缘。换H₂后检测到CH₂O和CH₃O中间体,CH₂O先升后降、CH₃O持续上升,表明连续加氢。DFT算出面内CO进一步加氢能垒太高(~1.89 eV),所以脱附溢流到边缘;边缘经*CHO→*CH₂O→*CH₃O→CH₃OH,决速步能垒1.42 eV。
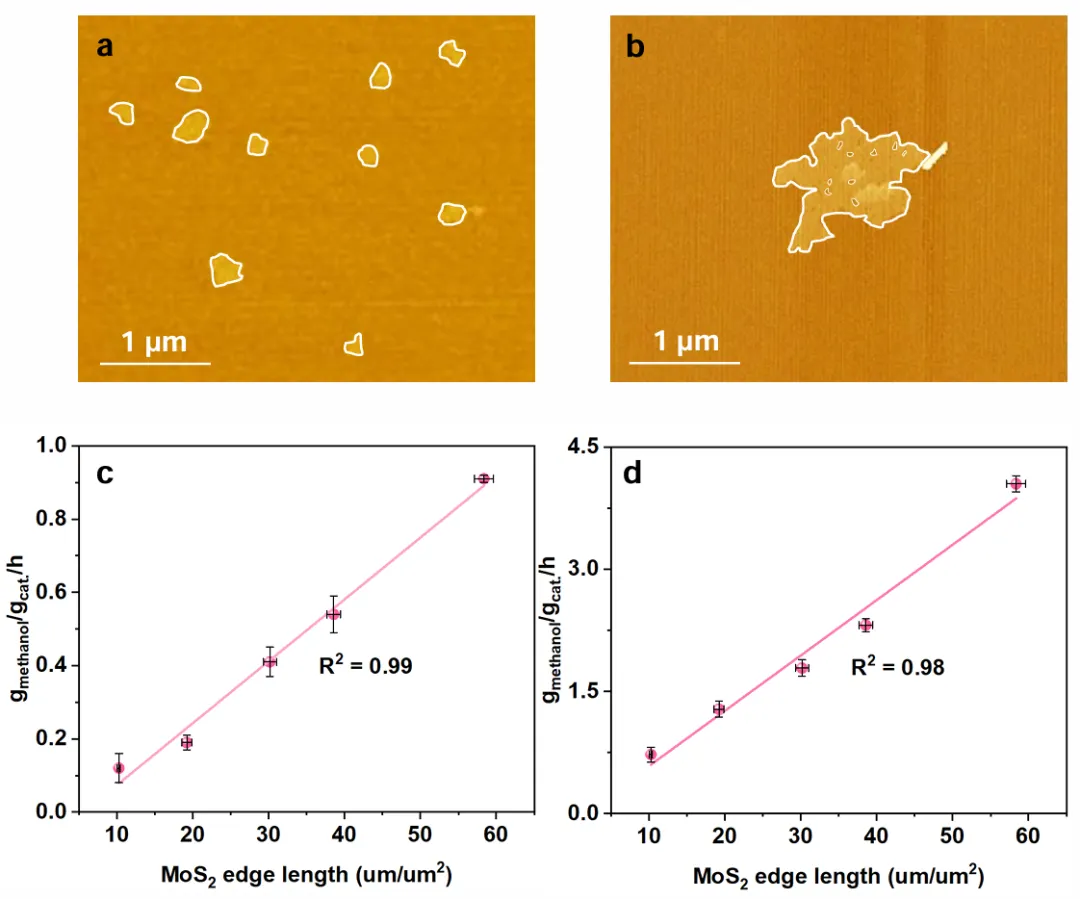
Figure 5验证边缘位点的重要性。样品在N₂中300°C热处理,边缘长度从58.4缩短到10.5 μm/μm²,甲醇速率和边缘长度正相关,但和比表面积无关;用CO直接做原料也观察到同样规律。
四、研究结论
说白了,作者靠1T′-MoS₂的二维结构,在同一片纳米片上同时搞定了CO₂解离和加氢两件事:面内的2OS位点把CO₂打断成CO,CO脱附后跑到边缘的EO-OS位点继续加氢,中间体传输效率比物理混合的体系高得多。研究把CO₂→*CO→*CH₂O→*CH₃O→CH₃OH整条路都摸清楚了,也解释清楚了为什么1T′相的氧化边缘能稳住C-O键、不让它断成甲烷。从动力学上看,材料对H₂几乎是零级反应,说明H₂不会堵活性位点;对CO₂是正级反应,CO₂可以持续活化。最终这个体系在210°C下做到了23.0%的转化率、99.2%的甲醇选择性和0.91 g/g/h的反应速率,循环五轮也没掉链子。这项工作不光给了一个好用的催化剂,更重要的是指了一条新路——通过相工程调控二维材料的边缘和缺陷,完全可以在单一材料里编排多步反应。
随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 在深圳,这4个地方的年轻人正在悄悄涌入
- 深圳“细胞十条”深度解读:200亿赛道的破局之道与产业新坐标
- 深圳市罗湖区黄贝街道茅台酒回收
- 文化润疆|第二十七届深圳读书月“文化润疆·深喀共读”系列活动之经典诗词赏析诵读会在喀什市举办
- 深圳大运天地随手拍
- 深圳办结全国首宗股票虚假陈述仲裁案入围2025年度深圳“十大法治事件”推荐事件
- 深圳1-6年级语文【第5单元资料】开始更新!
- 中国礼品家居“旗舰展”,第34届深圳礼品展参观指南来了→
- 《深圳市人力资源和社会保障局关于印发〈深圳市青年人才认定和管理办法〉的通知》的政策解读
- AI赋能·共话未来——我会参加深圳市湖南市州商会秘书长联席会AI主题交流活动
