深圳大学江智勇/张俊民Nature子刊 | 不对称催化分子内光环加成:对映选择性合成两类双环 [2.1.1] 己烷!
- 2026-06-18 12:44:47
深圳大学江智勇/张俊民Nature子刊 | 不对称催化分子内光环加成:对映选择性合成两类双环 [2.1.1] 己烷!
【做计算 找华算】华算科技预存增值高至30%,送¥8500+返利!Nature/Science课题组长期合作,全年科研无忧,快来抢占! 劲爆低价!DFT计算5折开抢!催化/电池/半导体计算全覆盖,服务至审稿意见结束! 邻二取代苯结构在药物和农药分子中广泛存在,近年来,化学家们对开发含有三维(3D)桥环骨架的其生物电子等排体表现出浓厚兴趣。 2026年4月9日,深圳大学张俊民、江智勇在国际知名期刊Nature Communications发表题为《Enantioselective Synthesis of 2-Oxabicyclo[2.1.1]hexanes and Bicyclo[2.1.1]hexanes via Catalytic Asymmetric Intramolecular Photocycloadditions》的研究论文,Dong Tian为论文第一作者,张俊民、江智勇为论文共同通讯作者。 
因此,4, 5-二取代-2-氧杂双环[2.1.1]己烷因其增强的药理活性、改善的代谢稳定性,以及尤为重要的优异水溶性,成为极具潜力的候选骨架。作者报道了通过光诱导能量转移(EnT)与手性布朗斯特酸协同催化的分子内[2+2]光环加成反应,实现该类化合物的对映选择性合成。以高产率、良好至优异的对映选择性和非对映选择性合成了一系列具有重要价值的吡啶官能化2-氧杂双环[2.1.1]己烷衍生物。 此外,该无过渡金属策略为构建吡啶基双环[2.1.1]己烷提供了高效且模块化的途径,此类化合物含有极具挑战性的结构单元——吡啶环直接连在桥头碳原子上。机理研究表明,光敏剂与低温反应对手性催化剂不可或缺地促进底物激发至三重态、进而在高活性转化中实现立体选择性控制起到关键作用。 在药物科学中,生物电子等排体设计已成为加速发现新药候选物的核心策略,能够使候选药物相比母体化合物具有更优的药理性质。在这些策略中,三维桥环骨架已成为苯环的有效生物电子等排体,而苯环是生物活性分子中最普遍的结构单元之一。特别是,鉴于邻二取代苯单元在300余种已上市药物和农药中的广泛存在,寻找其合适的生物电子等排体成为研究热点。因此,1, 5-二取代双环[2.1.1]己烷(BCHs)被认为是极具前景的替代骨架。 研究表明,将BCH骨架替代苯环能够增强关键分子性质,包括药理活性和代谢稳定性。然而,其水溶性与母体邻二取代苯相近,限制了该生物电子等排策略在药物设计中的更广泛应用。 2023年,Mykhailiuk及其团队解决了这一局限,提出4,5-二取代-2-氧杂双环[2.1.1]己烷作为更优的生物电子等排体,不仅保留理想的药理学特征,还表现出显著提升的水溶性。这类骨架可通过光诱导能量转移(EnT)催化过程从简单二烯化合物便捷制备。尽管如此,该方法的立体选择性中等(非对映选择性dr≤4:1)。更关键的是,尽管该类化合物拥有三个立体中心(平面邻二取代苯不具备的特征),但尚未实现从易修饰底物出发的对映选择性合成,而对映纯在手性药物开发中至关重要。这一难点可能源于三重态中间体的高反应性,阻碍了手性催化剂对其进行有效的对映面区分,同时也存在竞争性的外消旋背景反应。 值得注意的是,Bach及其团队提出了一种光激发策略以克服后一挑战,即底物2-(烯氧基)环己-2-烯酮的两个氧原子与手性铑催化剂原位形成的复合物,而非底物本身,在可见光下直接激发。此外,Zi及其团队报道了手性钯催化双环[1.1.0]丁烷(BCBs)与醛的分子间[2+2]环加成反应。然而,这些产物不具备邻二取代苯生物电子等排所需的结构特征,正如Mykhailiuk团队在比较不同取代模式的2-氧杂双环[2.1.1]己烷与对应邻二取代苯分子的生物活性时所证实的。因此,发展高效实用的对映选择性方法合成这类特定生物电子等排体仍是亟待解决的重要挑战。 毋庸置疑,简单二烯的分子内环加成反应凭借底物易得性以及优于分子间反应的化学与区域选择性,为合成具有多样4-位和5-位取代基的高值手性富集2-氧杂双环[2.1.1]己烷提供了极具前景的途径。近年来,该策略已成功用于构建其他苯环生物电子等排体。例如,Rigotti、Tortosa及其团队开发了一种光驱动不对称分子内[2+2]环加成反应,使用Λ-RhS作为手性路易斯酸催化剂,合成手性富集的5-酰胺官能化双环[2.1.1]己烷。该方法依赖Λ-RhS与二烯的酰胺部分相互作用形成光敏物种,在光照下产生三重态。为获得手性富集的2-氮杂双环[2.1.1]己烷(aza-BCHs),本团队发展了DPZ作为光敏剂、手性钪(III)配合物催化的不对称分子内[2+2]光环加成双催化体系。在此体系中,由于给电子胺取代基导致三重态烯烃反应性低,外消旋背景反应被抑制。手性钪(III)配合物作为路易斯酸提供足够的活化能以实现高效环化。这些发现表明,二烯底物中不同类型的烯烃可能需要不同的催化策略,这构成了重大挑战。 基于本团队在芳环生物电子等排体不对称光催化方法上的研究基础,本文报道了通过不对称分子内[2+2]光环加成反应合成手性富集4,5-二取代-2-氧杂双环[2.1.1]己烷的解决方案。采用无过渡金属双催化体系——手性布朗斯特酸与光敏剂协同能量转移(EnT)光催化,以高产率、良好至优异的对映选择性和非对映选择性合成一系列具有多样桥头吡啶取代基的产物。这类骨架作为临床和农用相关化合物的生物电子等排体,具有重要药学价值。关键的是,立体选择性控制依赖于光敏剂与手性布朗斯特酸的精准匹配。作为光敏剂的4CzIPN具有与底物烯烃相近的三重态能级,在低温下不经活化则无法反应。手性磷酸(CPA)通过轻微降低烯烃的三重态能级活化烯烃,使能量转移过程得以进行。这种协同作用在三个立体中心形成过程中实现高效对映面区分,同时抑制外消旋背景反应。重要的是,该方法的高效性与通用性实现了桥头含吡啶取代基的手性富集双环[2.1.1]己烷(BCHs)的精准合成。 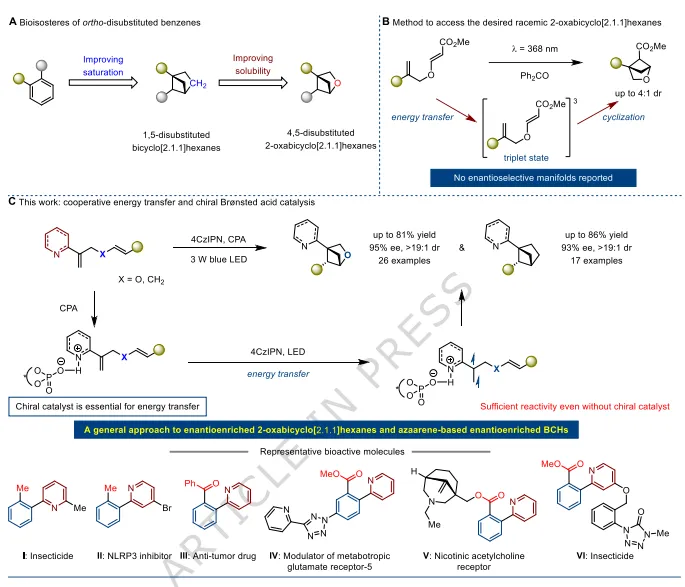
图1:研究背景与工作概述。(A)邻二取代苯的生物电子等排体发展:从双环[2.1.1]己烷到含氧杂环提升水溶性;(B)已有方法仅能得到外消旋2-氧杂双环[2.1.1]己烷,无不对称合成路线;(C)本工作:光敏剂+手性布朗斯特酸协同,实现[2+2]不对称光环化,构建手性杂环与双环骨架。 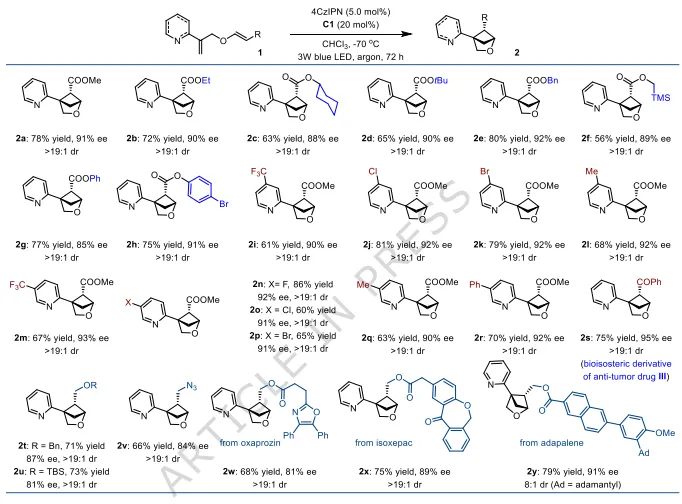
图2:2-氧杂双环[2.1.1]己烷底物拓展。涵盖不同酯基、吡啶环取代、烷基/官能化侧链、药物片段修饰底物;产物收率56-78%,ee=85-95%,非对映选择性均>19: 1,普适性优异。 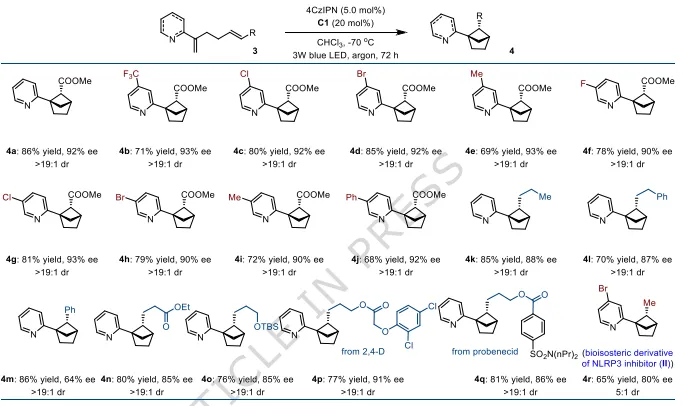
图3:吡啶基双环[2.1.1]己烷底物拓展。将氧换为碳,直接构建桥头连吡啶的手性双环[2.1.1]己烷;收率69–86%,ee64–93%,非对映选择性>19:1,可接入药物片段。 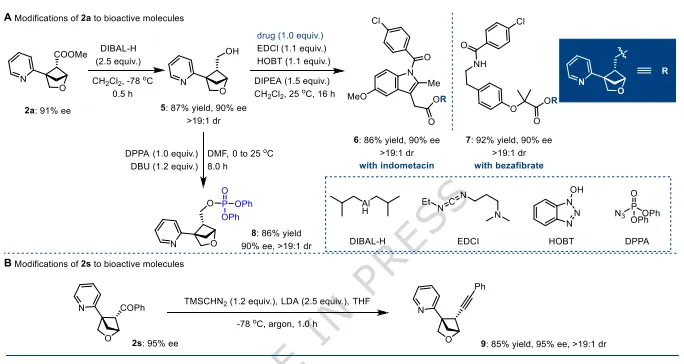
图4:产物衍生化(药用分子合成)。(A)2a经还原、酯化、磷酰化得到多种手性药物中间体,手性保持;(B)2s转化为炔基衍生物,可用于生物偶联。 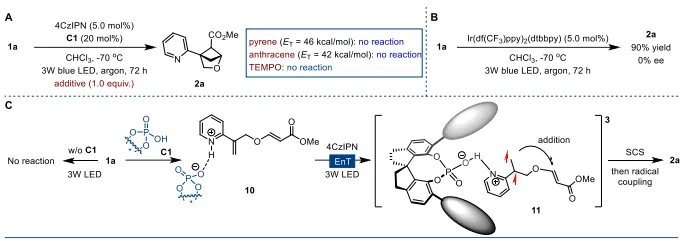
图5:机理研究。(A)淬灭实验证明为能量转移(EnT)、自由基中间体路径;(B)无手性酸时高温可反应但无选择性,低温仅手性催化路径进行;(C)机理:手性酸与底物形成离子对→降低三线态能量→光敏剂能量转移→手性环境下环化。 综上,作者发展了光诱导能量转移与手性布朗斯特酸协同催化体系,首次实现不对称分子内[2+2]光环加成反应,高效、高对映选择性构建2-氧杂双环[2.1.1]己烷与双环[2.1.1]己烷类化合物。在蓝光LED与低温条件下,产物收率优异,对映选择性最高达93%ee,非对映选择性>19:1,可兼容吡啶、酯、烷基、叠氮及多种药物片段。机理表明,光敏剂4CzIPN与底物三重态能量匹配,手性磷酸通过氢键调控实现立体选择性,抑制背景反应。该方法无需过渡金属,条件温和,可一步构建三个连续手性中心,产物可进一步衍生为醇、磷酸酯、炔烃等药物中间体。 该成果突破邻二取代苯生物等排体难以不对称合成的瓶颈,首次实现氧杂/碳桥双环己烷的高效手性构建,为饱和环烃生物等排体合成提供全新策略。产物作为苯环生物等排体,可显著改善药物溶解度、代谢稳定性与成药性,广泛适用于新药研发中骨架改造与分子设计,尤其适用于含吡啶片段的药物分子开发,对推动创新药物发现与绿色光合成化学发展具有重要价值。 Enantioselective Synthesis of 2-Oxabicyclo[2.1.1]hexanes and Bicyclo[2.1.1]hexanes via Catalytic Asymmetric Intramolecular Photocycloadditions. Nat. Commun., (2026). https://doi.org/10.1038/s41467-026-71590-x. #深圳大学#张俊民#江智勇#催化#Nature子刊 👉 点击阅读原文,立即下单!

本文来自网友投稿或网络内容,如有侵犯您的权益请联系我们删除,联系邮箱:wyl860211@qq.com 。

