Transl Psychiatry︱深圳大学沈立明团队揭示Ddx3x敲低通过突触可塑性异常诱导小鼠孤独症样行为
- 2026-06-01 01:30:49

撰文︱陈海毅
审阅︱沈立明
责编︱王思珍
孤独症谱系障碍(autism spectrum disorder,ASD)是一类常见的神经发育障碍疾病,其核心特征为社会交往与沟通缺陷,同时伴随重复刻板行为和兴趣狭窄等表现[1,2]。近年来,大量遗传学研究已鉴定出上百个ASD相关风险基因[3],但这些基因如何在特定脑区、特定细胞层面调控突触发育与神经环路功能,进而诱发ASD核心症状,仍缺乏系统性的机制阐释。
DDX3X是近年来备受关注的ASD核心风险基因之一[4,5],其编码的DEAD-box RNA 解旋酶参与RNA代谢、翻译调控、神经发生及突触形成等关键生理过程[6-8]。然而,DDX3X表达异常如何影响特定脑区功能并诱导ASD相关行为表型,其深层分子机制尚未明确。
近日,深圳大学生命与海洋科学学院沈立明教授团队在国际期刊Translational Psychiatry在线发表题为“Knockdown of Ddx3x in mPFC induces autistic-like phenotype in mice via altered synaptic plasticity”的研究论文。该研究通过构建HT22细胞Ddx3x敲低模型和小鼠内侧前额叶皮层(medial prefrontal cortex, mPFC)神经元特异性Ddx3x敲低模型,结合行为学、蛋白质组学、形态学和电生理学多维度分析(图1),系统揭示了DDX3X缺陷导致ASD样行为的潜在分子机制,提示突触可塑性异常和突触蛋白稳态失衡可能是其中的重要病理基础。(拓展阅读:沈立明课题组相关研究进展,详见“逻辑神经科学”报道(点击阅读):Mol Neurobiol︱深圳大学沈立明团队揭示接触蛋白4基因缺失小鼠出现孤独症样行为并伴肠脑轴代谢异常)

研究团队首先在小鼠海马神经元样HT22细胞中实现Ddx3x的特异性敲低,并验证了干预效率。实验发现,DDX3X表达下降后,HT22细胞的生长速度显著减慢,提示DDX3X对神经样细胞的正常增殖与生长具有不可或缺的作用。
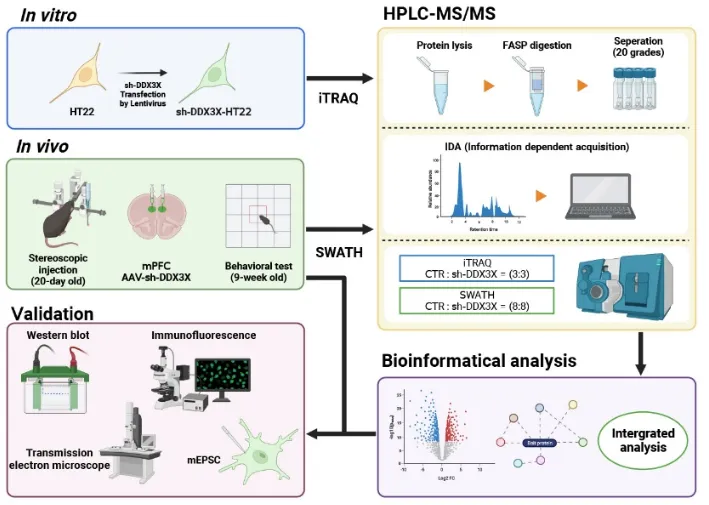
图1.研究整体实验流程图
为探究其分子变化,团队开展iTRAQ蛋白质组学分析,结果显示,Ddx3x敲低后出现大量差异表达蛋白,这些蛋白主要富集于蛋白折叠、tRNA代谢、高尔基体组织、伴侣蛋白依赖性蛋白重折叠,以及mTOR、ErbB等重要信号通路。该结果表明,DDX3X缺陷不仅直接影响细胞生长状态,还会引发细胞内广泛的蛋白质稳态失衡,为后续体内研究奠定分子基础(图2)。
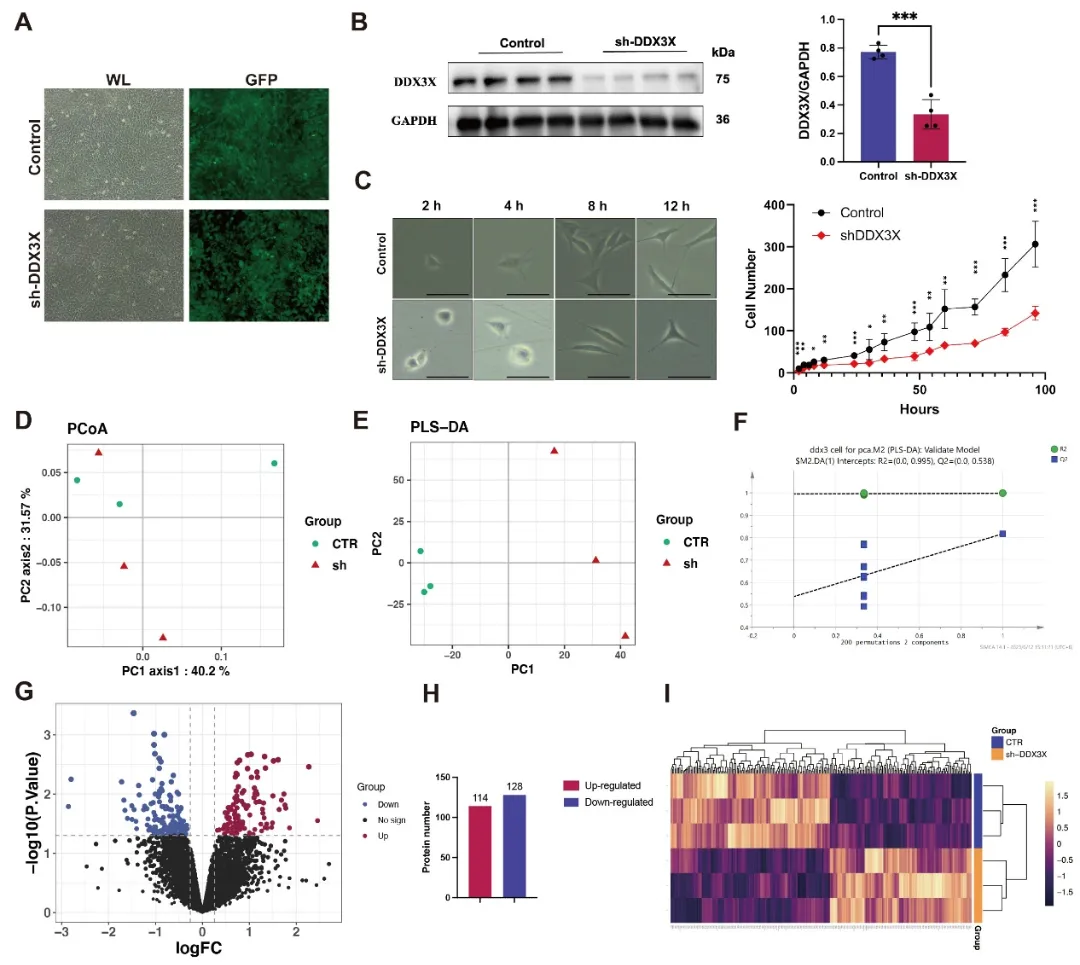
图2. HT22 细胞Ddx3x敲低验证及 iTRAQ 蛋白质组学分析结果
mPFC是调控社会行为、认知加工和记忆的核心脑区,与ASD核心症状的发生密切相关。研究团队利用AAV病毒在雄性小鼠mPFC双侧神经元中特异性敲低Ddx3x,成功构建体内疾病模型,Western blot和免疫荧光结果证实,模型小鼠mPFC内DDX3X蛋白表达显著下降,模型构建成功(图3A-E)。
随后,研究团队对模型小鼠开展系统的行为学检测。旷场实验结果显示,Ddx3x敲低未显著影响小鼠总移动距离、区域进入次数及边缘停留时间,说明模型小鼠的基础运动能力和焦虑样行为无明显异常,排除了非特异性因素的干扰(图3F, G)。
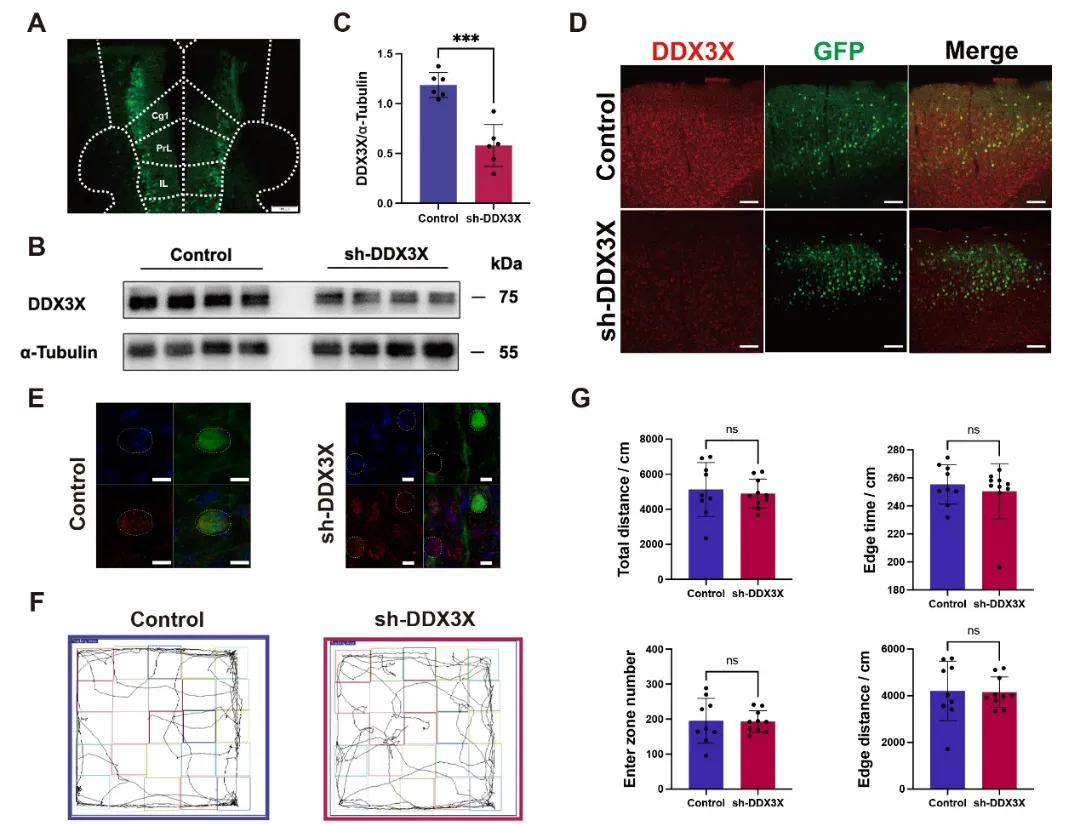 图3.小鼠mPFC内DDX3X敲低验证及旷场实验结果
图3.小鼠mPFC内DDX3X敲低验证及旷场实验结果
而在 ASD 核心行为检测中,模型小鼠表现出一系列典型的ASD样异常:三箱社交实验中,敲低组小鼠的社会偏好和社会新奇偏好均显著下降;Y迷宫实验显示,其在较长时间间隔下的空间识别记忆能力明显减弱;幼鼠社交互动实验中,肛生殖区嗅探等核心社交行为显著减少;弹珠掩埋实验中,敲低组小鼠的重复刻板行为则明显增强。上述结果表明,mPFC脑区内Ddx3x的下调,可诱导小鼠出现典型的ASD样行为表型(图4)。
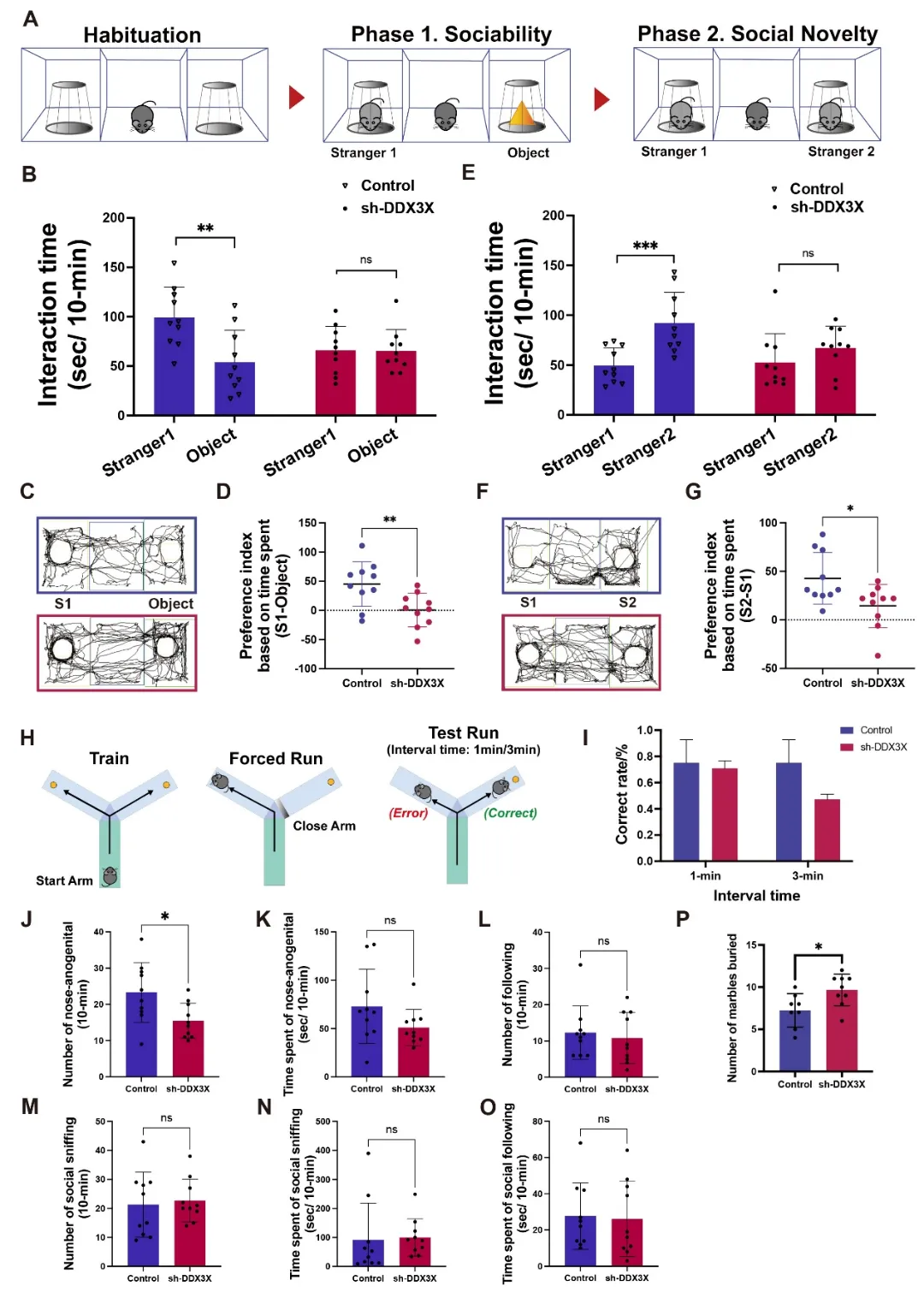
图4. 模型小鼠行为学检测结果(三箱社交、Y 迷宫、幼鼠社交互动、弹珠掩埋实验)
为解析mPFC区Ddx3x敲低诱导ASD样行为的分子机制,研究团队对小鼠 mPFC组织开展SWATH-MS高通量蛋白质组学分析,共鉴定出285个差异表达蛋白,且绝大多数表现为下调。
通路富集分析发现,这些差异蛋白主要富集于长时程增强(long-term potentiation, LTP)、谷氨酸能突触、GABA能突触、突触后致密区(postsynaptic density, PSD)、支链氨基酸降解及催产素信号通路等与突触功能密切相关的通路;进一步的SynGO突触特异性注释分析表明,差异蛋白显著富集于突触前、突触后及PSD相关结构,提示DDX3X缺陷对突触信号传递和突触结构维持产生了广泛且直接的影响(图5)。
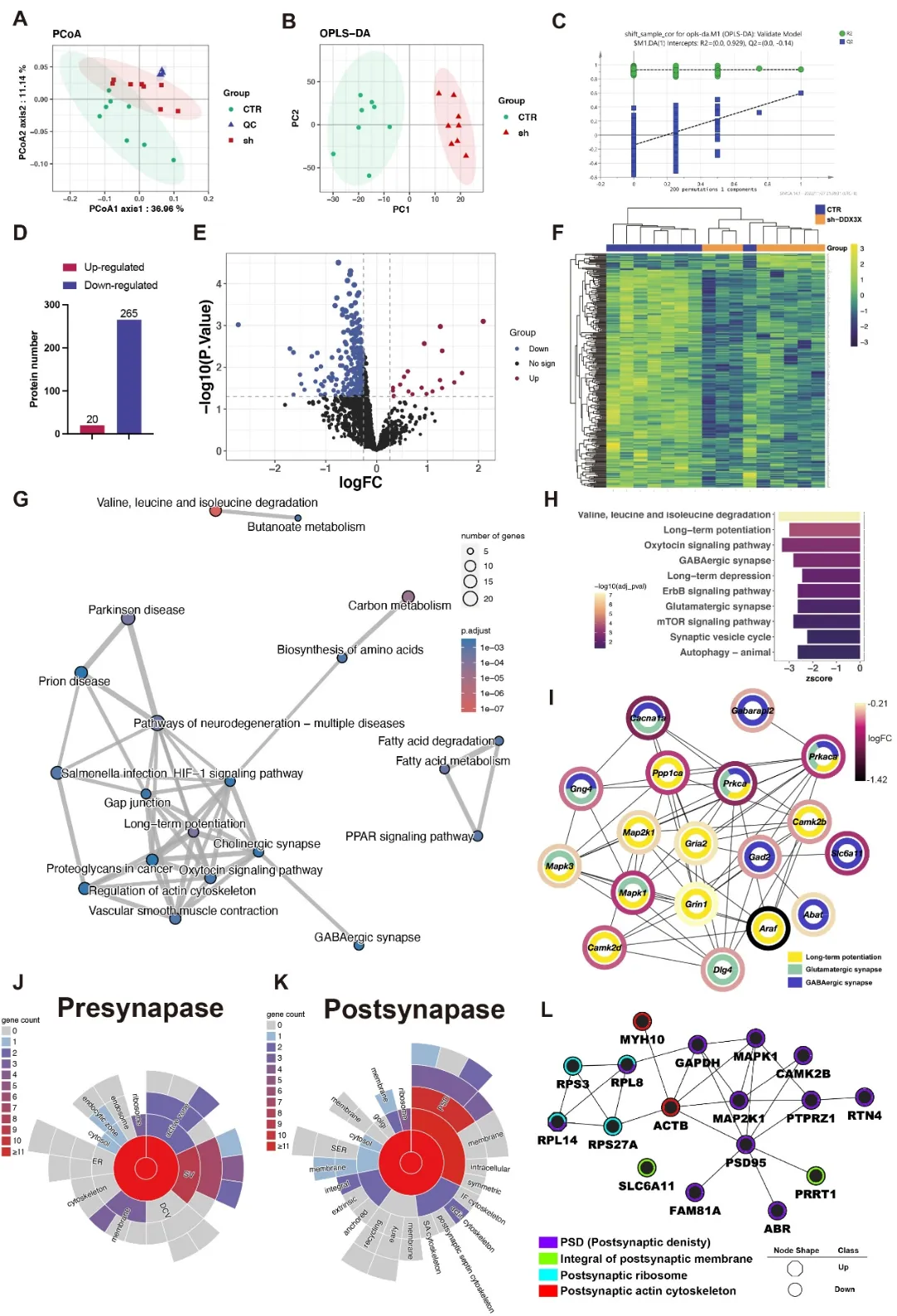
图5.小鼠mPFC组织蛋白质组学及通路富集分析结果
在此基础上,研究团队整合HT22细胞和小鼠mPFC的蛋白质组学数据,发现两种模型中存在共同受扰动的核心通路,其中最突出的为泛素-蛋白酶体系统(ubiquitin-proteasome system, UPS) 和从头蛋白折叠通路(de novo 蛋白折叠通路)。
已知UPS在蛋白降解、突触组成调节、神经元发育及突触修剪中发挥关键作用,而蛋白折叠异常则直接破坏细胞内蛋白质质量控制。该结果提示,DDX3X缺陷可通过同步抑制UPS和从头蛋白折叠通路,破坏突触蛋白稳态,进而加重神经元功能异常,这是DDX3X缺陷引发神经功能障碍的重要共性机制(图6)。
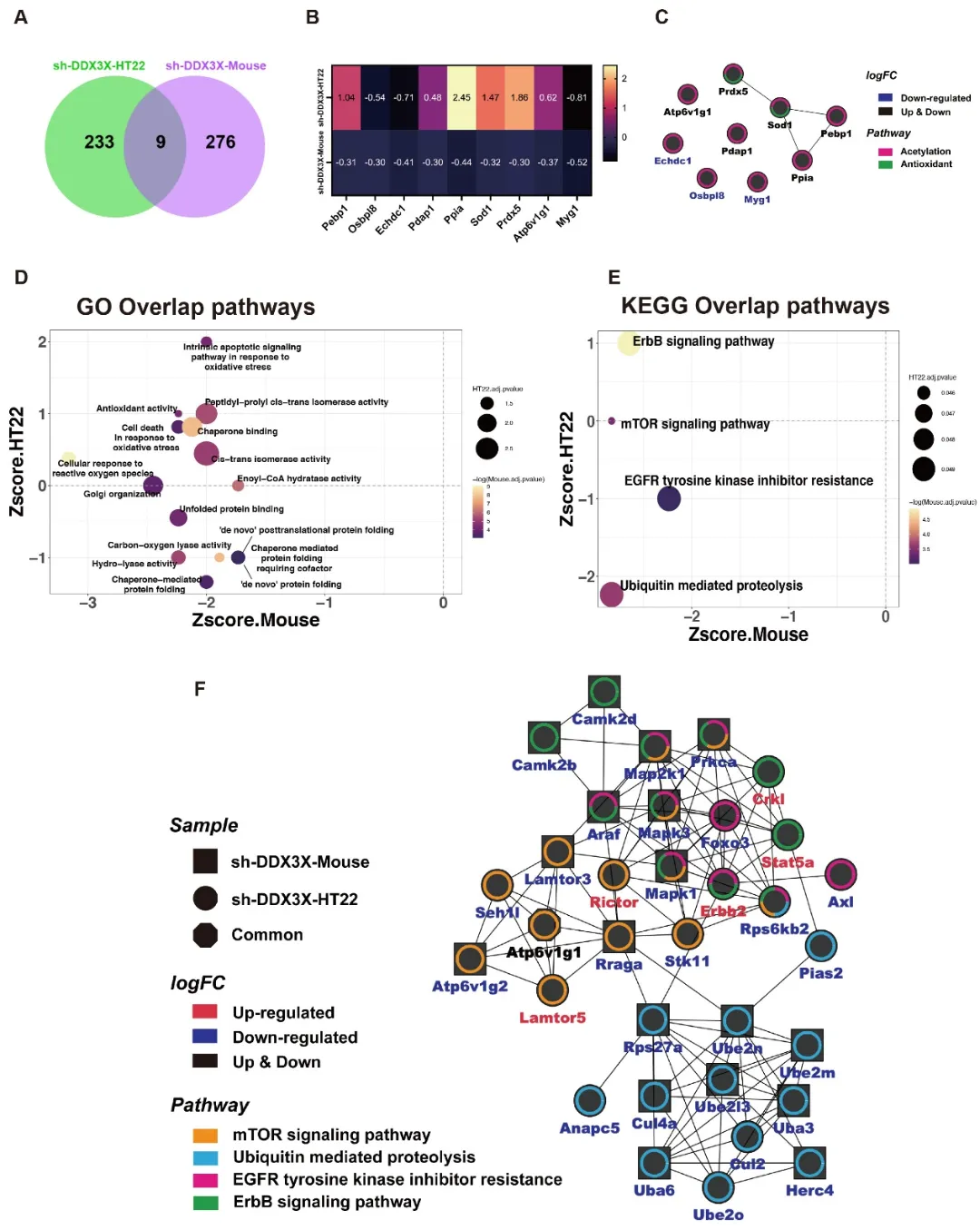
图6. HT22 细胞与小鼠mPFC组织联合蛋白质组学分析结果
为验证蛋白质组学结果,研究团队检测了多个与突触可塑性密切相关的关键蛋白,包括MAPK1、PKCA、PSD95、PRRT1、GRIA2、GRIN1和CAMKIID,结果发现这些蛋白在Ddx3x敲低组中均显著下调。其中,MAPK1、PKCA、CAMKIID 参与LTP相关信号转导,PSD95、PRRT1调控突触后支架结构与受体定位,GRIA2、GRIN1则为AMPA、NMDA谷氨酸受体核心成分,提示DDX3X缺陷通过影响突触后信号整合和支架稳定性,直接削弱突触可塑性。
在形态学层面,共聚焦显微成像显示,Ddx3x敲低后小鼠mPFC神经元树突棘总密度显著下降,其中短粗型(stubby)和蘑菇型(mushroom)等成熟树突棘减少更为明显,而细长型(thin)树突棘无显著变化;透射电镜结果进一步显示,敲低组小鼠兴奋性突触的PSD厚度和面积明显减小,PSD长度和突触间隙宽度则无明显改变,证实DDX3X缺陷主要影响突触后致密区的成熟与稳定。
在功能层面,膜片钳电生理记录显示,模型小鼠mPFC神经元的微小兴奋性突触后电流(mEPSCs)频率显著下降,而振幅无明显变化。mEPSC频率下降提示功能性兴奋性突触数目减少或突触释放事件发生率降低,振幅不变则说明单个突触后反应强度未受影响。结合树突棘减少和PSD结构异常的结果,证实DDX3X缺陷可导致兴奋性突触数量和成熟度下降,进而显著削弱mPFC内的突触传递功能(图7)。
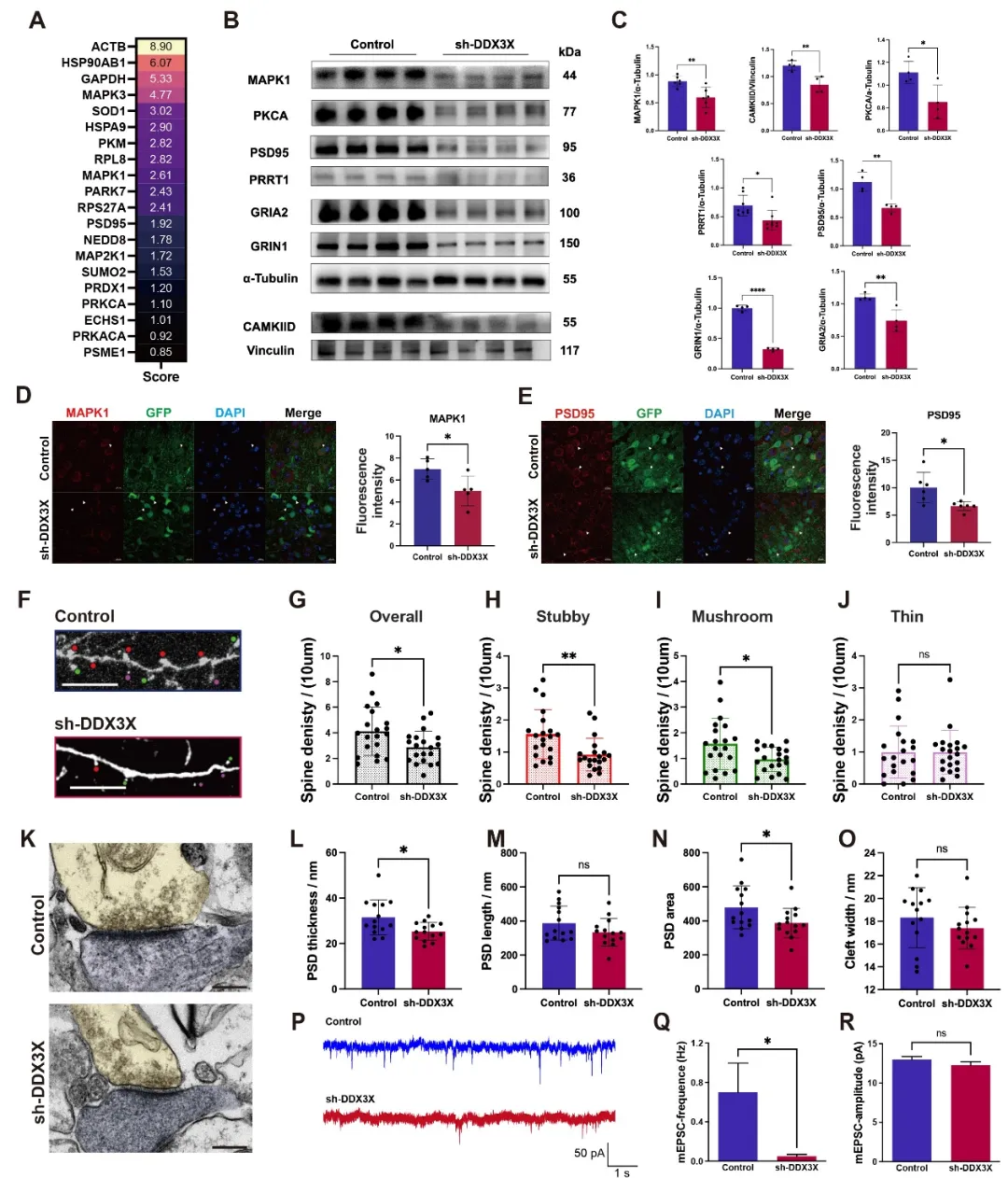
图7.突触可塑性关键蛋白验证、树突棘形态分析及电生理学检测结果
综上所述,本研究结果表明:小鼠mPFC神经元中Ddx3x的特异性下调,可直接诱导其出现典型的 ASD 样行为表型,且该过程伴随突触可塑性相关蛋白广泛下调、成熟树突棘减少、PSD结构异常及兴奋性突触传递减弱。
从机制上看,DDX3X缺陷不仅直接抑制LTP、谷氨酸能/GABA能突触等经典突触功能通路,还可通过同步抑制UPS和从头蛋白折叠通路,破坏突触蛋白稳态,最终导致神经元功能障碍和ASD样行为。该研究将DDX3X风险基因、mPFC脑区功能、突触可塑性损伤与ASD样行为表型进行系统关联,为理解DDX3X相关ASD的发病机制提供了全新的实验证据。
同时,研究团队也指出了本研究的局限性:目前尚缺乏DDX3X恢复表达的救援实验,DDX3X缺陷与行为异常之间的因果关系仍需进一步强化;研究主要聚焦于神经元,对星形胶质细胞、小胶质细胞等非神经元细胞中DDX3X的作用尚未展开;雌性小鼠及海马、杏仁核等其他ASD相关脑区的作用也有待后续探究。
未来,若能结合条件性恢复表达、跨细胞类型分析及多脑区比较研究,将有助于进一步阐明DDX3X调控神经发育和突触稳态的具体分子机制,为DDX3X相关神经发育障碍的靶向干预提供新的理论依据和潜在靶点。
原文链接:https://www.nature.com/articles/s41398-026-03981-z
深圳大学生命与海洋科学学院沈立明教授为论文通讯作者,硕士研究生庄鸿彬和博士后曹雪杉为论文共同第一作者。
通讯作者简介:沈立明,深圳大学生命与海洋科学学院教授,主要研究方向为疾病多组学,聚焦于孤独症、阿尔茨海默症等神经精神疾病诊断生物标志物及发病机制的研究。在国内外高水平期刊发表研究相关学术论文100余篇,参编中英文专著4部,研究成果获教育部自然科学二等奖1项。获批国家发明专利8项、PCT专利2项。
本研究获得了国家自然科学基金、深圳市科技创新委员会、深港脑科学创新研究院及深圳大学研究生创新培养基金的资助。
转载须知:“逻辑神经科学”特邀稿件,且作者授权发布;本内容著作权归作者和“逻辑神经科学”共同所有;欢迎个人转发分享,未经授权禁止转载,违者必究。

“逻辑神经科学”微信群:文献学习

[1] Lawrence KE, Hernandez LM, Fuster E, Padgaonkar NT, Patterson G, Jung J, et al. Impact of autism genetic risk on brain connectivity: a mechanism for the female protective effect. Brain. 2022,145:378-87.
[2] Ellingford RA, Panasiuk MJ, de Meritens ER, Shaunak R, Naybour L, Browne L, et al. Cell-type-specific synaptic imbalance and disrupted homeostatic plasticity in cortical circuits of ASD-associated Chd8 haploinsufficient mice. Mol Psychiatry. 2021, 26:3614-24.
[3] Havdahl A, Niarchou M, Starnawska A, Uddin M, van der Merwe C, Warrier V. Genetic contributions to autism spectrum disorder. Psychological Medicine. 2021, 51:2260-73.
[4] Levy T, Siper PM, Lerman B, Halpern D, Zweifach J, Belani P, et al. DDX3X Syndrome: Summary of Findings and Recommendations for Evaluation and Care. Pediatr Neurol. 2023,138:87-94.
[5] Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014, 515:216-21.
[6] Duan W, Huang G, Sui Y, Wang K, Yu Y, Chu X, et al. Deficiency of DDX3X results in neurogenesis defects and abnormal behaviors via dysfunction of the Notch signaling. Proc Natl Acad Sci U S A. 2024, 121:e2404173121.
[7] Chen CY, Chan CH, Chen CM, Tsai YS, Tsai TY, Wu Lee YH, et al. Targeted inactivation of murine Ddx3x: essential roles of Ddx3x in placentation and embryogenesis. Hum Mol Genet. 2016, 25:2905-22.
[8] Lennox AL, Hoye ML, Jiang R, Johnson-Kerner BL, Suit LA, Venkataramanan S, et al. Pathogenic DDX3X Mutations Impair RNA Metabolism and Neurogenesis during Fetal Cortical Development. Neuron. 2020, 106:404-20.e8.


