科研│中国热科院&深圳基因所&浙江农科院:整合基因组学、转录组学和代谢组学揭示了花烛属花部性状的遗传结构
- 2026-04-16 05:42:44
 点击蓝字↑↑↑“转录组”,轻松关注不迷路
点击蓝字↑↑↑“转录组”,轻松关注不迷路
 生科云网址:https://www.bioincloud.tech
生科云网址:https://www.bioincloud.tech论文ID
实验设计
结果
1 两个花烛属物种的高质量基因组组装与注释
本研究获得了两个高质量染色体水平的花烛属(Anthurium)基因组。红掌(Anthuriumandraeanum “Alabama”)包含15条染色体,总长度为4.15 Gb,支架N50值为219.78 Mb;火鹤花(A. scherzerianum “Red Lantern”)总长度为3.84 Gb,支架N50值为242.70 Mb,与K-mer分析结果一致(表1,图1B,图S1-S3)。研究者使用BUSCO程序收集的1416个保守胚胎植物蛋白评估组装完整性,显示红掌(A. andraeanum)和火鹤花(A. scherzerianum)的完整性分别为97.6%和98.1%(表S4,图S4)。两个基因组均表现出较高的碱基准确性、覆盖度和组装质量(表S5)。Hi-C染色质接触图显示了强烈的染色体内相互作用和清晰的染色体间边界,证实了组装的准确性和染色体水平的连续性(图S5A和S5B)。此外,研究者比较分析表明,基于Hi-C的组装与来自花烛(A. andraeanum)双亲分离群体的高密度遗传图谱高度一致(图S5C和S5D)。总之,这两个新的基因组组装表现出优异的连续性、准确性和完整性。
研究者在红掌(A. andraeanum)和火鹤花(A. scherzerianum)基因组中分别预测到3.41 Gb和3.15 Gb的转座元件(TE)序列,分别占组装总长度的82.07%和81.96%(表S6)。在TE中,Gypsy元件占主导地位,平均占每个组装注释TE含量的42.06%,占总组装数量的34.50%(表S6)。研究者通过整合从头预测、同源性预测和转录组证据预测,在红掌(A. andraeanum)和火鹤花(A. scherzerianum)中分别预测到52,380和61,287个蛋白质编码基因,分别覆盖了96.3%和96.4%的BUSCO基因(表S7,图S4)。平均而言,火鹤花(A. scherzerianum)的总基因长度比红掌(A. andraeanum)长,而火鹤花(A. scherzerianum)的平均内含子长度更长(表S7)。此外,研究者分别在红掌(A. andraeanum)和火鹤花(A. scherzerianum)基因组中注释了1993和2826个非编码RNA(ncRNA),包括175和134个microRNA(miRNA)、653和1030个转运RNA(tRNA)、564和1074个核糖体RNA(rRNA)以及597和583个小核RNA(snRNA)(表S7)。
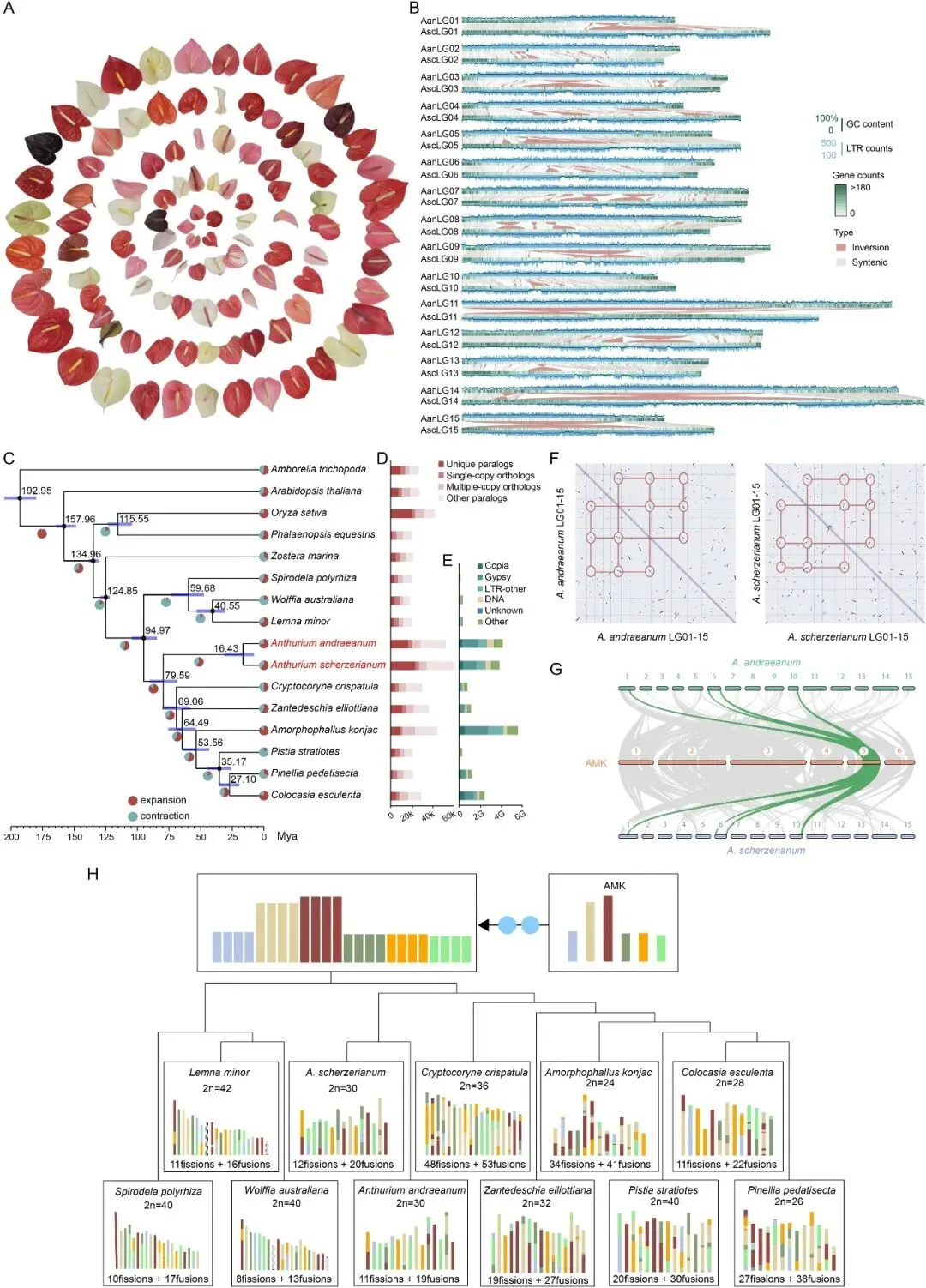
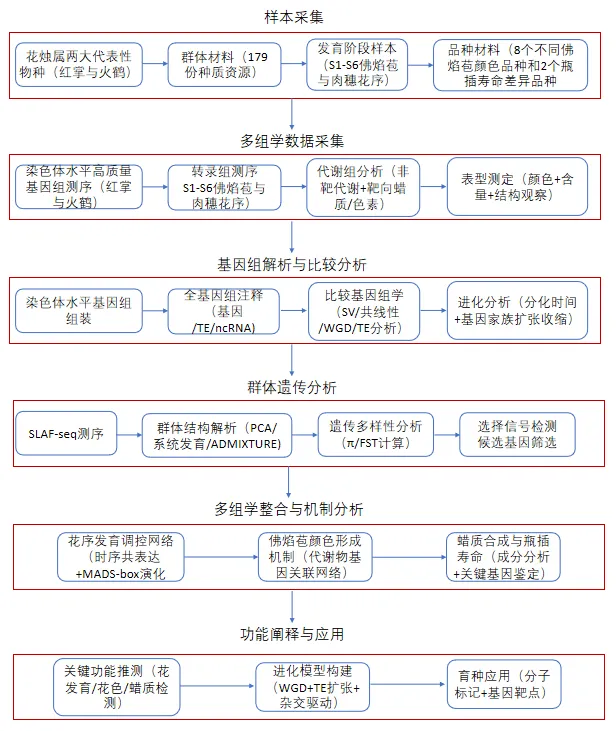
图1. 比较基因组学分析。(A)花烛属佛焰苞的多样性。(B)红掌(A. andraeanum)和火鹤花(A. scherzerianum)的基因组特征及两个花烛属基因组之间的结构变异分布。(C)时间校准的系统发育树,标注了节点年龄和95%置信区间。饼图显示了经历扩张或收缩的基因家族比例。柱状图显示了基因家族类别和转座元件含量。黑点表示分析中使用的化石校准点。(D)基因家族分类。(E)11个天南星科物种重复序列含量的比较。花烛属物种和A. konjac的基因组中,DNA转座子和LTR逆转录转座子的长度远大于其他物种,这与它们较大的基因组大小密切相关。(F)两个花烛属基因组内的内部比较。两个花烛属基因组的宏观共线性模式显示,每个区域与自身内部的四个共线性区域对齐。(G)两个花烛属基因组与单子叶植物祖先核型的共线性比较。清晰的4:1共线性关系支持了两个花烛属物种中发生两次全基因组复制事件。(H)天南星科基因组从单子叶植物祖先核型开始的进化情景。虚线表示的染色体代表未被单子叶植物祖先核型覆盖的区域。比较核型分析揭示,天南星科基因组经历了广泛的染色体重排,导致了谱系特异性的基因组结构。
2 进化与比较基因组学分析
研究者以红掌(A. andraeanum)为参考基因组,在火鹤花(A. scherzerianum)中鉴定出3950万个单核苷酸多态性(SNP)和384万个小的插入缺失(InDel,长度<50 bp),其中13号和3号染色体每千碱基的SNP和InDel数量最高(表S9)。在这些变异中,715808个SNP和52666个InDel位于编码序列区域(表S10)。相对于红掌(A. andraeanum)基因组,共检测到2727045个结构变异(SV),包括1078798个缺失、1564762个插入、10935个易位、725个倒位和71825个重复(图1B,表S11)。值得注意的是,在除2、4、6、10和13号染色体外的大多数染色体上观察到了兆碱基规模的倒位和易位,这些染色体显示出更多的结构变异(图1B,表S11)。这些广泛的序列和结构变异可能导致了两个花烛属物种之间观察到的形态、生理和生态分化。
基于来自16个物种的441个单拷贝直系同源基因构建了具有分化时间估计的最大似然系统发育树,包括本研究生成的两个花烛属物种,以及来自天南星科其他属的9个物种,并以大叶藻(Zostera marina)、小兰屿蝴蝶兰(Phalaenopsis equestris)、水稻(Oryza sativa)、拟南芥(Arabidopsis thaliana)及无油樟(Amborella trichopoda)作为外群物种。花烛属与其最近亲缘类群的分化时间约为7959万年前(Mya),而红掌(A. andraeanum)和火鹤花(A. scherzerianum)之间的分化时间估计约为1643万年前(图1C)。研究者分别在红掌(A. andraeanum)和火鹤花(A. scherzerianum)基因组中鉴定出9148和16041个独特的旁系同源基因(图1D)。独特旁系同源基因的KEGG富集结果表明,它们主要分布在“角质、木栓质和蜡质生物合成”、“脂肪酸生物合成”、“卟啉和叶绿素代谢”以及“脂质代谢”途径中,这表明花烛属中与代谢物合成和环境适应途径相关的关键酶基因发生了进化(图S6)。在红掌(A. andraeanum)和火鹤花(A. scherzerianum)基因组中,分别有1454和1131个基因家族显著扩张,594和611个基因家族收缩(图1C)。扩张的基因家族富集在"苯丙烷生物合成"、"玉米素生物合成"和"植物-病原体互作"途径,这些途径与花烛属适应环境的植物耐受性和抗逆性相关(图S7)。通过比较11个天南星科物种中各种重复序列的含量,研究者观察到两个花烛属物种和A.konjac基因组中的DNA转座子和LTR逆转录转座子(包括Copia、Gypsy等元件)的长度远大于其他八个天南星科物种,这与基因组大小的扩张密切相关(图1E,表S6)。研究者对两个花烛属基因组中LTR插入时间的分析揭示了Copia(红掌A. andraeanum:18千年;火鹤花A. scherzerianum:16千年)和Gypsy(红掌A. andraeanum:20千年;火鹤花A. scherzerianum:8千年)元件在两个谱系中不同的爆发事件(图S8)。红掌(A. andraeanum)和火鹤花(A. scherzerianum)中的Copia扩张事件发生在一个狭窄的时间范围内,而Gypsy活性在谱系间表现出显著差异。这些发现表明,谱系特异性的调控机制或环境压力塑造了TE的动态。
全基因组复制(WGD)事件被广泛认为是物种形成的主要驱动力,提供了丰富的遗传物质,促进了物种多样化并增强了环境适应性,从而在植物进化过程中发挥着关键作用。保守同线性区块的鉴定是推断WGD事件的基础。基因组内共线性分析在每个基因组内发现了广泛的复制区块,支持了红掌(A. andraeanum)和火鹤花(A. scherzerianum)中发生两次WGD的假说(图1F)。与重建的单子叶植物祖先核型(AMK)的比较揭示了清晰的4:1同线性关系,为两个花烛属物种中发生的两次WGD事件提供了有力支持(图1G,图S9;表S12和S13)。Ks分布分析显示了一个与古老WGD事件相关的单峰(图S10)。虽然Ks分布没有显示出两个明显分离的峰,但这种模式与其他天南星科物种的先前报告一致,其中时间接近或重叠的WGD事件也导致了单一的Ks峰。这些观察结果共同支持了天南星科谱系中发生两次WGD事件,尽管仅使用Ks分布可能难以解析。此外,染色体重排事件被认为是基因组进化和物种形成的重要因素。基于AMK,研究者对天南星科14个选定物种的进化轨迹分析显示,浮萍亚科的核型保持相对稳定(图S11)。相比之下,其余的天南星科物种经历了复杂的染色体融合和结构重排,导致现存物种的染色体数目(2n = 24–40)和核型模式高度多样化(图1H)。基于AMK的天南星科(Araceae)物种核型进化分析,揭示了染色体变异如何驱动该科内的物种多样化。
3 种群结构分析
为了更好地了解多样化个体内的动态基因组变异和多样性,研究者根据园艺应用和地理分布,选择了总共179份花烛属(Anthurium)材料,分为4类——切花(CF)、盆花(PF)、观叶盆栽植物(OFP)和观叶花烛(OFA)(图2A和B,表S14)。通过特异性位点扩增片段测序(SLAF-seq)生成了总共22.6亿条reads,平均碱基识别准确度Q30达到94.70%,平均GC含量为41.31%。研究者使用BWA软件将clean reads比对到红掌(A. andraeanum)参考基因组。个体对参考基因组的比对率在80.33%到99.41%之间,表明179份花烛属材料与红掌(A. andraeanum)基因组具有高度的遗传相似性。其中,来自荷兰的“Choco”显示出最高的比对率(99.41%),而来自中国西双版纳的H22254比对率最低(80.33%)(表S14)。随后,研究者鉴定出14006947个高质量SNP,并从这个SNP数据集中筛选出1055个4DTv位点用于后续的群体遗传学分析。
基于高质量SNP的主成分分析(PCA)显示,179份花烛属(Anthurium)材料聚类成两个主要的遗传组:簇I(包括CF、PF和OFA材料)和簇II(包括CF、PF、OFA和OFP)(图2C)。基于相同SNP数据集的系统发育分析得出一致的分类,将材料分为两个主要分支(图2C和D;图S12A)。此外,在K = 3时的群体结构分析证实了这种划分,清晰地勾勒出两个组群,与PCA和系统发育结果一致(图2C - E,图S12B)。簇I和簇II之间的全基因组配对FST估计为0.17,表明存在中等程度的遗传分化。簇I的核苷酸多样性(π)为1.26 × 10-5,簇II为1.98 × 10-5,表明两组都保持着大量的组内遗传变异(图2F)。与这些结果一致,基于SNP的聚类并不完全支持传统的CF、PF、OFP和OFA园艺分组,表明这些分类只部分反映了潜在的遗传结构。
通过比较两个簇,基于π簇I与π簇II比率的前5%区域被确定为选择性清除热点,这些区域与基因注释相交得到26个候选基因,其中大多数功能未知。值得注意的是,有4个基因已被深入研究:AanV1_01G014960(RHF2A),参与配子发生和减数分裂后有丝分裂;AanV1_05G028290(WRKY13),调控茎发育并增强镉耐受性;AanV1_06G027080(ARF19),通过调节生长素和乙烯响应基因的表达来控制根和茎的发育;以及AanV1_07G016990(RPE),卡尔文-本森循环中影响光合能力的关键酶。类似地,Fst分析将前5%的区域确定为分化热点,得到16个功能未知的候选基因。总之,这些热点区域的候选基因可能为花烛属的性状形成和适应性进化提供了分子基础。
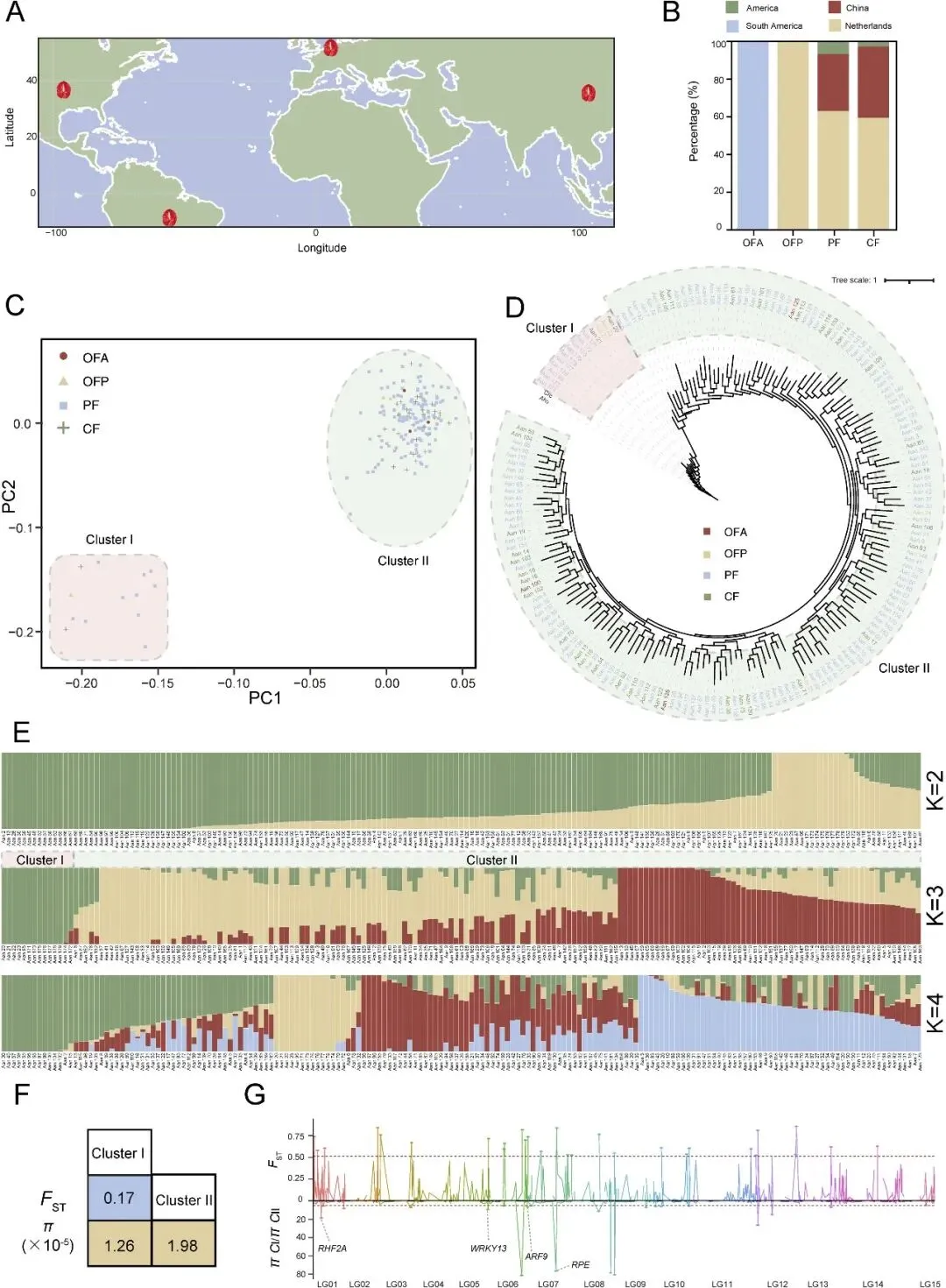
图2. 花烛属(Anthurium)材料的地理分布、遗传多样性和群体结构。(A)本研究中使用的179份花烛属材料的全球分布,显示其地理来源。(B)各园艺组别(切花、盆花、观叶盆栽植物和观叶花烛)内不同地理区域的比例。(C)基于高质量SNP对179份花烛属材料进行的主成分分析,揭示了两个主要遗传簇。(D)基于1000次自展重抽样的最大似然系统发育树,显示两个主要分支。每个节点显示1000次重复的自展值,表示分支支持水平。材料聚集成两个主要分支,与主成分分析和群体结构分析结果一致。(E)基于SNP数据集对179份花烛属材料进行的群体结构分析。每个垂直条代表一份材料,颜色表示分配给不同簇的遗传组分比例,与系统发育和主成分分析结果一致。(F)每个簇内的平均核苷酸多样性以及簇间的群体分化。(G)花烛属材料簇I与簇II之间的全基因组选择性清除筛选。红色虚线分别代表前5% FST值和前5% (π簇I/π簇II) 比率的阈值。
4 天南星科花序发育的调控动态
为了分析红掌(A. andraeanum)肉穗花序发育的调控,研究者将任意两个阶段之间的差异表达基因(DEG)输入时序基因共表达网络(TO-GCN)分析(图3A)。主要的GCN包含10个时序层级(图3B中表示为L1至L10),与六个开花阶段(S1–S6)中转录因子(TF)基因的表达时间顺序相匹配。总共发现10178个基因(650个TF和9528个结构基因),其平均TPM大于0.5,并且在六个开花时间点中任意两个样本间表现出显著分化。作为初始节点,研究者选择了在第一个时间点高表达但后期不表达的MADS-box转录因子APETALA1(API)来生成TO-GCN。TO-GCN揭示了一个涉及650个差异表达TF和15个花发育ABC模型基因的共表达网络(图3C)。总体模式显示,大多数网络成员(166个TF)出现在S1–S3,82个TF出现在S4–S5,28个TF出现在S6(图3B和C)。
为了理解花烛属花序相较于其他物种的进化基础,研究者选择了14个天南星科物种(包括本研究中的两个花烛属物种)和一个近缘物种(大叶藻Z. marina)进行进一步分析。基于佛焰苞和花序形态,这14个天南星科物种可分为三类(图3D)。第I类以未修饰的佛焰苞和单性花序为特征,包括紫萍(Spirodela polyrhiza)、澳州无根萍(Wolffia australiana)以及浮萍属(Lemna)物种。第II类具有扩展的平面佛焰苞和单性花序,包括红掌(A. andraeanum)和火鹤花(A. scherzerianum)。第III类以扩展的平面佛焰苞和两性花花序为特征,包括皱波隐棒花(Cryptocoryne crispatula)、马蹄莲(Zantedeschia elliottiana)、A.konjac、大薸(Pistia stratiotes)、水芋(Cryptocoryneescuienta)和虎掌(Pinellia pedatisecta)。此外,研究者基于14个天南星科物种的蛋白质序列构建了系统发育树,并观察到与上述类型相似的分组模式(图3D)。通过分析15个物种中MADS-box基因的拷贝数,鉴定出物种特异性的丢失、扩张和收缩(图3D,表S15)。研究者发现SOC1同源基因在第I类中缺失。相反,E功能基因AGL6同源基因在第III类中缺失。值得注意的是,两个花烛属物种同时保留了SOC1和AGL6同源基因。II型MADS-box基因的系统发育分析揭示了一个独特的进化枝与13个已建立亚组并存,这些基因被称为物种特异性基因(属于II型但未落入任何已建立的13个亚组)。这些基因在13个天南星科物种中发现,除了紫萍(Spirodela polyrhiza)。值得注意的是,所研究的两个花烛属物种拥有最多的物种特异性基因(图3E,图S13,表S16)。这可能与它们在花形态多样化中的中间进化位置相关,突显了它们在天南星科进化轨迹中的独特地位。
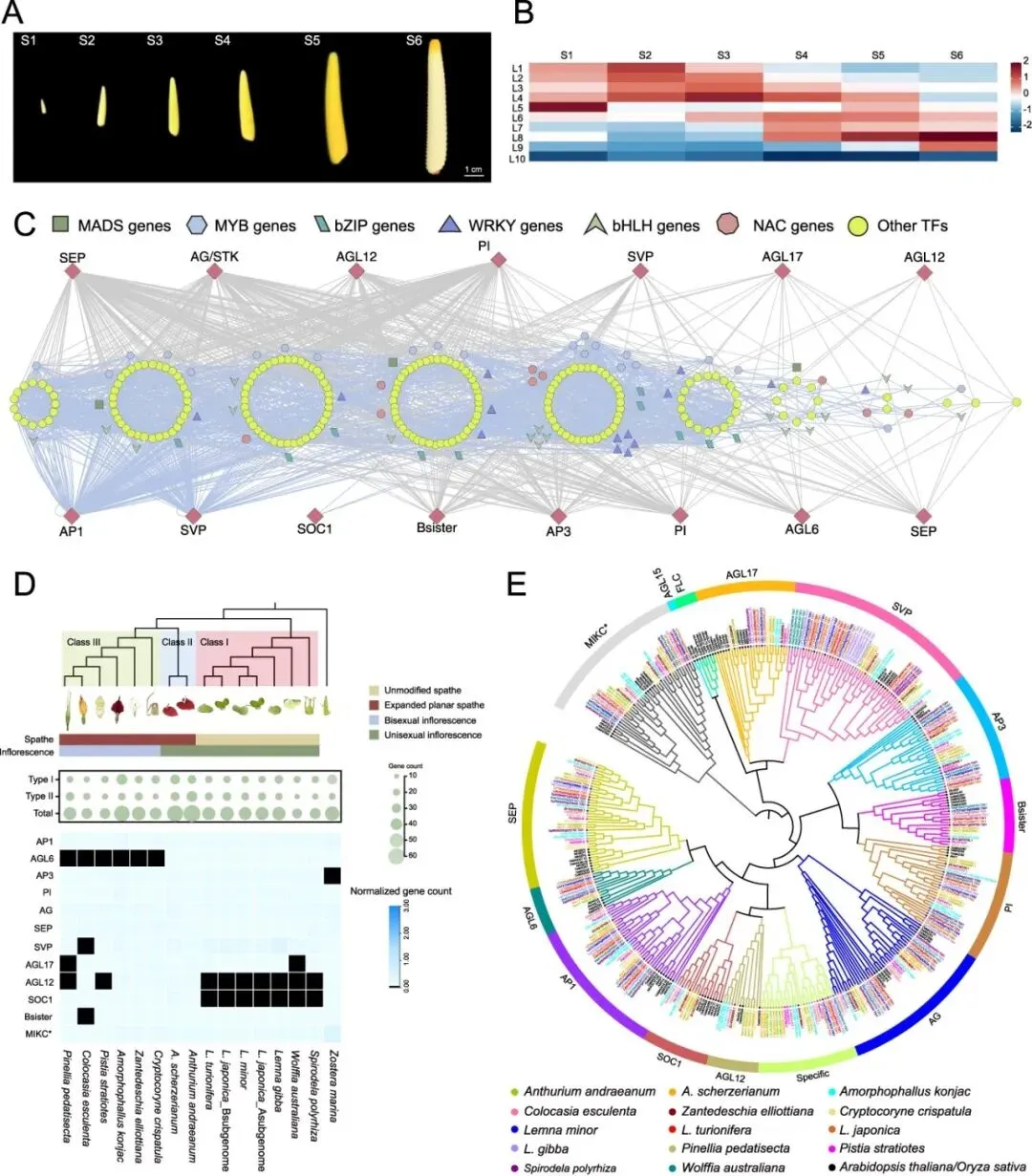
图3. 花烛属花序发育调控的动态。(A)花烛的六个肉穗花序发育阶段。(B)花烛各肉穗花序发育时间点上,时序基因共表达网络每个水平的平均TPM热图。(C)预测的调控网络以及参与花发育途径的转录因子和MADS基因之间的联系。L1到L10表示在有序基因共表达网络中识别的水平。(D)天南星科物种花序形态和标准化基因拷贝数的比较。对科数据集进行归一化,将每个物种的基因计数除以其科内最大的基因拷贝数。(E)天南星科物种MADS基因家族的最大似然系统发育树。
5 花烛属佛焰苞颜色形成的调控网络
佛焰苞颜色是花烛属的一个关键观赏性状。为了研究佛焰苞着色的调控网络,研究者鉴定了类黄酮(尤其是花青素)生物合成中的关键基因,并分析了它们在佛焰苞发育过程中的表达模式以及花青素的积累(图4A,图S14)。在佛焰苞发育过程中,类黄酮含量呈现双相振荡模式,从S1到S3阶段初始下降,随后在S5阶段逐渐上升,接着在S6阶段显著下降至最低点(图S15A)。相比之下,花青素积累呈现单向轨迹,在发育阶段中稳步增加,并在S5阶段达到峰值,这与佛焰苞完全着色在时间上相关(图S15B)。研究者进行了时序比较转录组分析,以探索影响佛焰苞发育过程中色素沉积的调控网络。基于与佛焰苞着色相关的表达模式,时序亚网络可分为四种主要模式:L1-L4在S1阶段高表达,L5-L8在S2阶段高表达,L9-L11在S3阶段高表达,以及L1-L11在S4-S6阶段普遍低表达(图4B)。在S1-S3阶段,许多TF,包括ERF、bHLH、bZIP、GRAS、MADS、MYB、NAC和WRKY,高表达(图4C)。这表明与类黄酮和花青素生物合成相关的基因主要在这些阶段被激活并发挥积极的调控作用。相比之下,在S4-S6阶段,活跃转录的TF基因数量显著下降,只有少数家族,如bHLH、MYB、TCP和NAC,仍然活跃(图4C)。
此外,基于红掌(A. andraeanum)的高质量基因组组装,研究者鉴定了所有与类黄酮和花青素生物合成相关的基因。研究者鉴定了33个在花青素和类黄酮生物合成中具有预测功能的酶基因(图4D)。研究者进行了Pearson相关性分析以评估关键酶基因的表达谱与花青素和类黄酮含量之间的关系。除了黄酮醇合酶(FLS),每个剩余关键酶基因的至少一个拷贝与花青素或类黄酮含量呈显著正相关(相关系数 > 0.8,p < 0.05;图4D)。这些结果支持了这些基因在类黄酮生物合成中的正向调控作用。此外,基于花烛(A. andraeanum)佛焰苞发育(S1–S6)过程中的基因表达模式(L1–L11)构建了TO-GCN(图4C),突出了33个酶基因与TF之间的显著相关性。相关性模式显示,酶基因与TF之间的关联主要分布在L1–L6,主要涉及查尔酮异构酶(CHI)、类黄酮3'-羟化酶(F3'H)、花青素合酶(ANS)、4-香豆酰辅酶A连接酶(4CL)、查尔酮合酶(CHS)和二氢黄酮醇还原酶(DFR)等酶基因(图4C)。总体而言,这些调控网络为进一步分析红掌(A. andraeanum)佛焰苞空间着色的分子机制提供了参考。
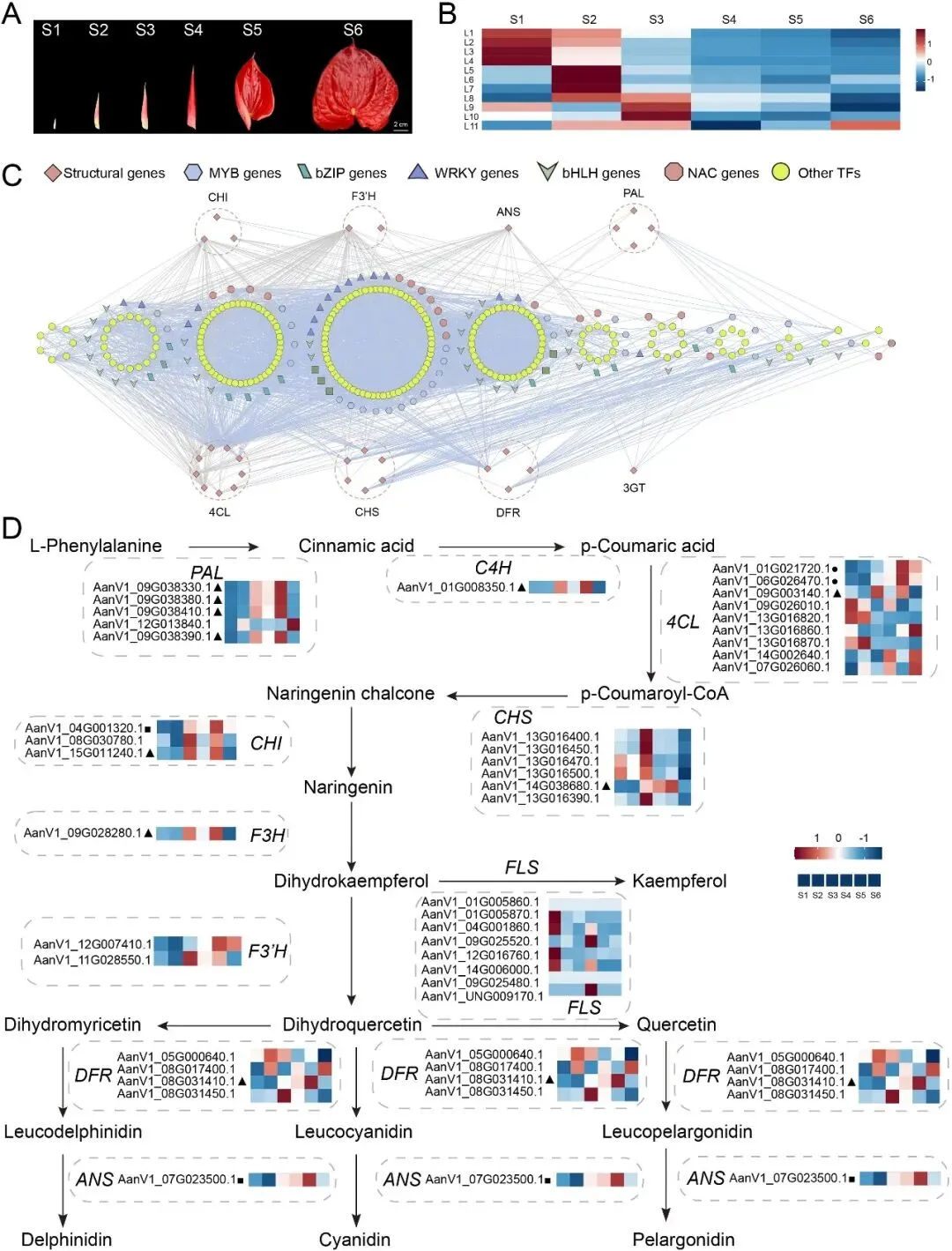
图4. 花烛属佛焰苞着色调控的动态。(A)红掌(A. andraeanum)的六个佛焰苞发育阶段。(B)花烛各佛焰苞发育时间点上,时序基因共表达网络每个水平的平均TPM热图。(C)预测的调控网络以及参与花青素/黄酮醇生物合成途径的转录因子和结构基因之间的联系。L1到L11表示在有序基因共表达网络中识别的水平。(D)花烛中的花青素/黄酮醇生物合成途径。与花青素和/或类黄酮含量显著正相关的基因(皮尔逊相关系数 > 0.8,p< 0.05)用颜色编码:红色代表花青素,蓝色代表类黄酮,绿色代表两者。
6 代谢物分析揭示不同代谢物群驱动的佛焰苞颜色多样性
为了阐明花烛属品种颜色调控的机制,研究者检测了八个具有不同佛焰苞颜色的品种(图5A)。颜色值被量化并在散点图中可视化,展示了不同品种间的颜色变化(图5B)。然后,研究者进行了代谢组学分析以研究佛焰苞颜色与代谢物组成之间的潜在关系。基于代谢物谱的聚类显示,白色和粉色佛焰苞紧密聚在一起,而橙色和紫色佛焰苞之间也观察到类似的紧密关联(图5C)。此外,研究者计算了总类黄酮含量,发现以白色佛焰苞为参照,红色、粉色和紫色佛焰苞的类黄酮含量显著更高(图5D)。研究者使用非靶向代谢物分析,鉴定出10种属于天竺葵素(天竺葵素衍生物)和矢车菊素(矢车菊素和芍药素衍生物)的花青素(图5E)。然而,在所有品种中仅鉴定出四种花青素。红色和黑色佛焰苞积累了大多数花青素(10种中的8种)的较高含量,而白色、绿色和粉色佛焰苞的所检测花青素含量相对较低。中间热图中的类黄酮呈现出与花青素相似的模式(图5E)。研究者进一步将佛焰苞颜色的参数与代谢物谱相关联,以探索佛焰苞颜色形成的可能机制。基于相关性的网络显示,L*a*b值与不同代谢物群之间的比率呈负相关(图5F,表S17)。例如,a与14个比率参数相关,包括类胡萝卜素与花青素的比率、类胡萝卜素与类黄酮的比率、花青素与类黄酮的比率,以及类胡萝卜素和花青素之和与类黄酮的比率。除了a,b仅与比率相关参数相关。D参数与大多数类胡萝卜素代谢物类别相关。这些结果表明,佛焰苞的感官颜色可能源于不同代谢物类别的复杂调控。
由于在非靶向代谢物分析中鉴定出花青素和类胡萝卜素,研究者筛选了负责花青素和类胡萝卜素生物合成的候选基因(图5G和H)。相关性热图突出了与花青素具有较强关联的候选基因(图5G),显示AanV1_07G004120、AanV1_13G016860、AanV1_07G023500、AanV1_01G008350、AanV1_04G001320、AanV1_13G016450、AanV1_08G031410、AanV1_09G028280、AanV1_12G007410和AanV1_09G038330分别是3-O-糖基转移酶(3GT)、4CL、ANS、C4H等关键酶的候选基因。根据相关性模式,类胡萝卜素生物合成中的关键基因有多个候选基因参与(图5H)。例如,八氢番茄红素合酶(CrtB)的AanV1_09G030060和AanV1_UNG028520均与类胡萝卜素强相关。LUT1也观察到与CrtB相似的模式(图5H),表明需要进一步的鉴定来筛选候选基因。
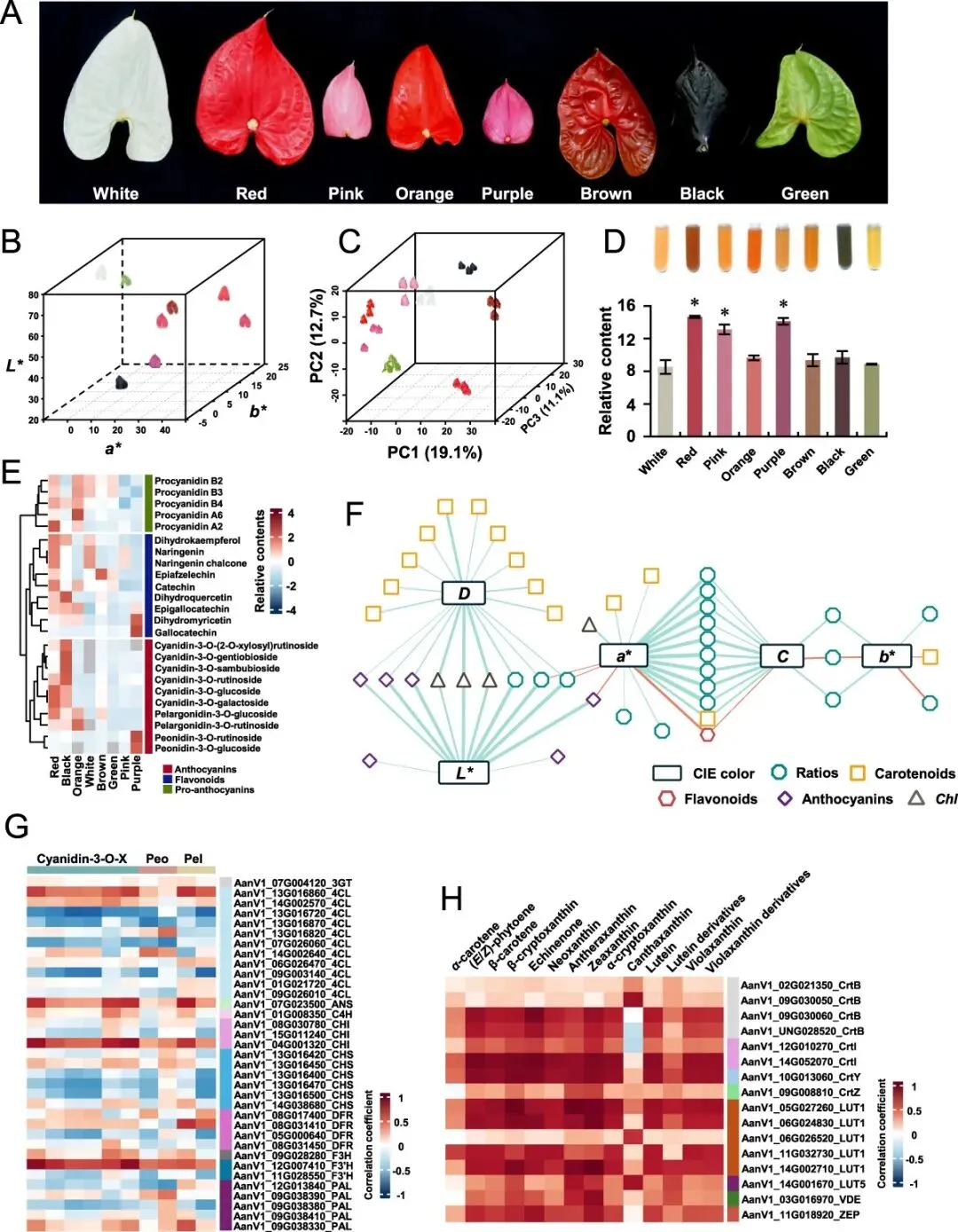
图5. 代谢物分析揭示不同代谢物群驱动的佛焰苞颜色形成。(A)不同品种苞片的图像。(B)Lab色彩空间中颜色差异的3D散点图。(C)非靶向代谢物分析的主成分分析图。(D)从八个品种佛焰苞中提取的总类黄酮。(E)花青素、类黄酮和原花青素的相对含量。*表示使用Dunnett检验,白色品种与其他品种之间存在显著差异。(F)Lab颜色性状与不同代谢物群之间基于相关性的网络。(G)候选基因与花青素之间相关性的热图。Cyanidin-3-O-X、Peo和Pel分别代表图中所示的矢车菊素、芍药素和天竺葵素。(H)候选基因与类胡萝卜素之间相关性的热图。
7 蜡质层延长作为观赏植物的花烛佛焰苞瓶插寿命
为了探索可能延长花烛瓶插寿命的角质层蜡质调控机制,研究者选择了两个瓶插寿命不同的红掌(A. andraeanum)品种“Alabama”和“Xavia”。具有更长保鲜期的“Alabama”,其佛焰苞表面比“Xavia”更亮,表明两个品种间佛焰苞的角质层蜡质存在差异。研究者使用扫描电子显微镜检查了两个品种佛焰苞上的角质层蜡晶体(图6A)。可以清楚地观察到颗粒状蜡晶体,其数量从“Alabama”的S3阶段开始显著增加。在“Xavia”的表皮上稀疏地观察到颗粒状蜡晶体(图6A,第1和第2行)。除了蜡晶体数量,蜡晶体和佛焰苞的横截面也显示出类似的迹象,即在S3之后,“Alabama”表现出比“Xavia”更清晰的蜡层(图6A,第3和第4行)。为了探索“Alabama”和“Xavia”之间蜡质差异的潜在机制,研究者分析了两个品种的角质层蜡,并鉴定出82种与角质层蜡相关的代谢物。在这些代谢物中,烷烃是主要的代谢物类别(表S19),表明红掌佛焰苞的角质层蜡主要由烷烃组成。PCA图显示,两个品种的早期阶段(S1和S2)与其他四个阶段明显分离(图6B),反映了发育阶段间代谢物组成的差异。与佛焰苞横截面观察结果一致(图6A),蜡沉积在前两个阶段似乎不太显著。此外,由于后期阶段(S3和S4)彼此接近,两个品种可能呈现出相似的蜡质谱。聚焦于蜡质谱,两个品种在大多数代谢物的积累趋势上没有差异(图6B)。值得注意的是,有七种代谢物在两个品种间的含量和积累模式上存在显著差异,表明这些代谢物可能在两个品种角质层蜡的变异中起作用(图6C)。研究者鉴定了三个参与蜡质生物合成和转运的基因(CER3、KCS1和KCS3),并获得了它们在两个品种不同发育阶段的表达水平(图6D)。CER3和KCS1基因的表达模式非常相似,而在两个品种的佛焰苞中,KCS3基因的表达水平存在明显差异,在“Alabama”中呈上升趋势,在“Xavia”中呈下降趋势(图6D)。研究者进一步探讨了已鉴定基因与蜡质谱之间的关联,以阐明角质层蜡形成的可能机制(图6E)。基于相关性的网络显示,三个基因KCS1、KCS3和CER3与几种代谢物显著相关。CER3的表达与Meta 73和二十九烷的含量呈正相关(图6E),并且在“Alabama”和“Xavia”之间存在显著差异(表S19)。
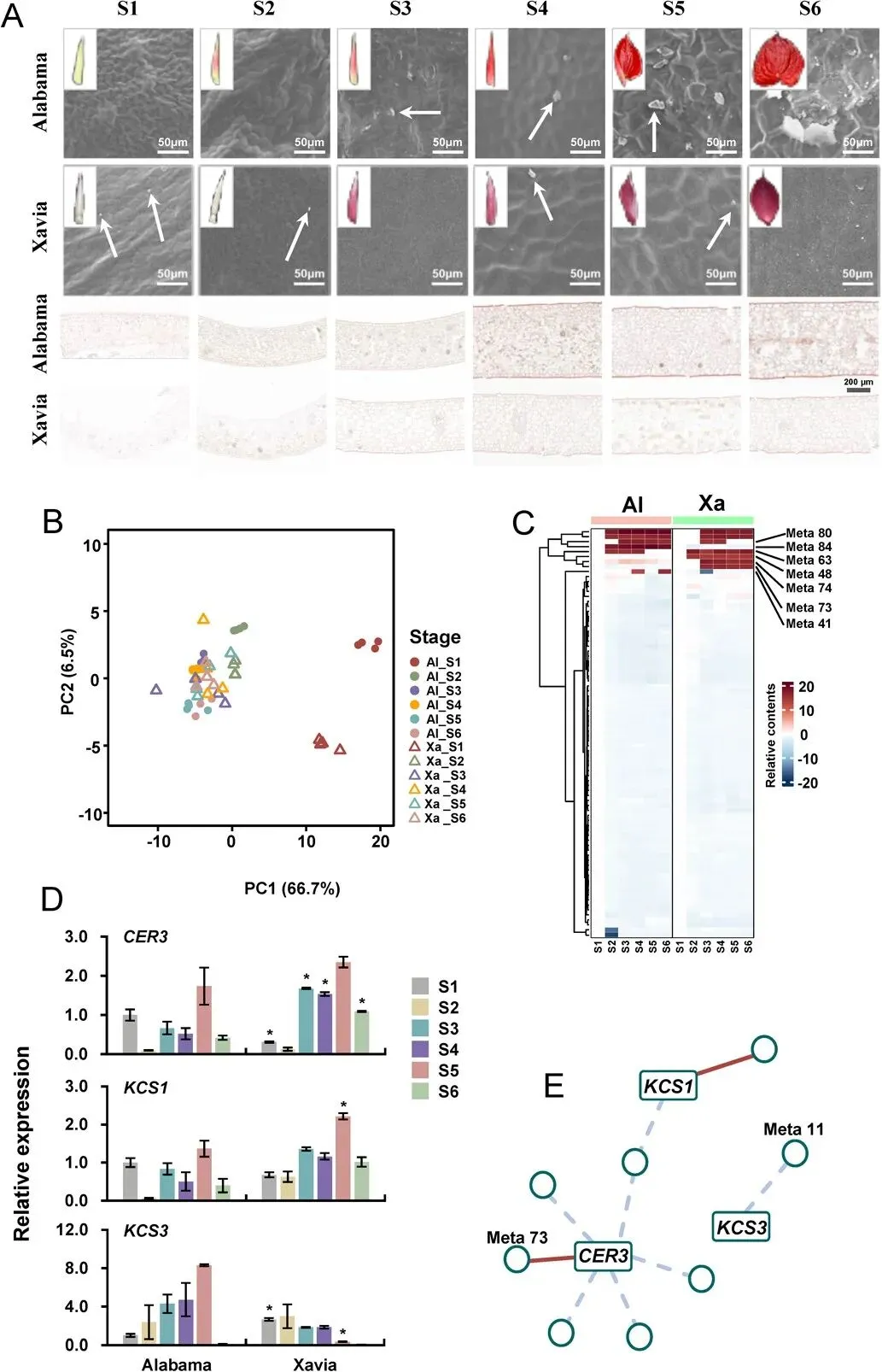
图6.“Alabama”和“Xavia”佛焰苞发育过程中的角质层蜡质。(A)分别使用扫描电子显微镜和冷冻切片分析佛焰苞表面的晶体和佛焰苞横截面的蜡层。箭头指示蜡晶体。左上角的照片显示了不同发育阶段的佛焰苞。(B)“Alabama”和“Xavia”在不同发育阶段佛焰苞中蜡相关代谢物的主成分分析图。(C)“Alabama”和“Xavia”在不同发育阶段蜡相关代谢物的相对含量。(D) )“Alabama”和“Xavia”佛焰苞中蜡质生物合成相关候选基因的表达水平。*表示使用t检验,')“Alabama”和“Xavia”在不同发育阶段存在显著差异。(E)代谢物与候选基因之间的相关性分析。
讨论
在本研究中,研究者展示了两个最知名的花烛属物种(红掌A. andraeanum和火鹤花A. scherzerianum)的染色体水平基因组组装,并进行了全面的比较基因组学分析,以阐明它们的基因组分歧、进化轨迹和适应机制。群体结构分析揭示了花烛属种群间复杂的遗传混合,这是由广泛的历史杂交驱动的。通过整合基因组学、转录组学和代谢组学数据集,研究者揭示了关键花部性状(特别是肉穗花序的进化和佛焰苞颜色的多样化)的遗传基础。
比较基因组学分析揭示了两个花烛属物种之间显著的结构差异,标志性特征是大规模的染色体重排。基因家族分析鉴定出谱系特异性扩张,特别是与苯丙烷生物合成、胁迫响应和病原体防御相关的基因家族,这可能为花烛属在多样化热带环境中的生态适应提供了分子基础。此外,TE活性,特别是Gypsy和Copia等LTR逆转录转座子内的活性,在基因组大小变异和结构进化中发挥了关键作用。Copia元件经历了相对同步的扩增,而Gypsy元件的扩增时间线则更为分散。这些TE扩增事件在时间上与末次冰期剧烈的气候变化相吻合,表明可能存在关联。然而,重要的是要注意,时间上的相关性本身并不能证实TE激活的适应性作用。需要进一步的功能验证来确定TE介导的基因组可塑性是否促进了冰期后的环境适应。尽管如此,这种基因组灵活性理论上可以促进对环境变化的快速适应,并可能驱动表型分化,从而加速花烛属的物种形成和性状多样化。鉴定出两次时间接近的WGD事件,这与在其他天南星科物种中观察到的模式一致。虽然这些共享的WGD反映了保守的进化机制,但花烛属基因组显示出独特的加速结构重组和TE增殖轨迹,与A.konjac的基因组扩张相似,但与紫萍的简化基因组形成对比。这些发现强调了保守的多倍化事件和谱系特异性的基因组创新如何共同塑造天南星科的生态适应,推进了研究该科进化驱动力的比较框架。然而,尽管鉴定出广泛的基因组变异,研究者对这些差异如何贡献于物种特异性性状的理解仍然受到缺乏系统表型和功能数据的限制。许多变异发生在编码区域内或涉及大规模重排,可能影响基因调控和表达。未来的整合研究,结合转录组学分析、定量表型分析和功能验证,对于阐明花烛属中的基因型-表型关系以及驱动物种多样化的分子机制至关重要。
本研究的全基因组分析鉴定出花烛属中两个主要的遗传群体,得到了PCA、系统发育和群体结构分析的一致支持。然而,这两个群体之间0.17的中等全基因组配对FST值表明存在部分遗传分化,同时伴随着持续的基因流。广泛的混合很可能是由反复的杂交事件导致的,而育种过程中相对较弱的人工选择使得多样的遗传背景得以保留。重要的是要认识到杂交和人工选择是不同的进化过程:杂交促进了基因混合,而选择则减少了目标位点的多样性。两个群体内分别为1.26 × 10-5和1.98 × 10-5的π值进一步支持了群体内存在大量变异。研究者通过π和FST分析鉴定的热点区域提示了潜在的选择目标。在26个基于π和16个基于FST的候选基因中,大多数功能未知,但有四个特征明确的基因——RHF2A、WRKY13、ARF19和RPE——突出了可能受选择的关键生物过程:RHF2A在配子发生和减数分裂后有丝分裂中发挥作用,表明生殖发育是潜在的选择目标;WRKY13和ARF19参与茎发育和激素响应性生长调控,可能影响植物结构和发育;RPE是卡尔文-本森循环中的关键酶,表明光合效率也可能受到适应性选择。总的来说,这些结果表明,花烛属中的选择可能作用于多个功能途径,包括繁殖、结构生长和光合作用,这有助于生态适应和重要的观赏性状。类似地,弱遗传聚类和普遍基因流模式在其他天南星科物种中也有报道,如水芋(Colocasia esculenta)和A.konjac,其中驯化和杂交塑造了群体结构。相比之下,紫萍(S. polyrhiza)表现出更强的与生态特化相关的遗传分化,突显了天南星科内不同的进化力量。与表现出更清晰的驯化驱动分化的物种不同,花烛属弱的种群结构可能反映了低强度人工选择和反复杂交繁殖的共同作用。这些发现强调了传统基于性状的园艺分类的局限性,并突显了利用先进基因组学工具阐明驯化历史和指导育种的必要性。虽然广泛的混合使得定义离散的遗传群体变得复杂,并可能降低性状关联研究的效力,但它同时反映了丰富的遗传多样性,这对育种和保护工作具有重要价值。
MADS-box基因家族对花发育至关重要,包括SOCI和AGL6等关键调控因子。研究者对14个天南星科物种中ABCDE模型基因的比较分析揭示了与适应性创新相关的谱系特异性丢失模式。SOCI的丢失与原始佛焰苞的进化相关,可能影响花序结构并增强生态位适应。相反,AGL6的丢失与第III类谱系中单性花的进化同时发生,表明其可能参与花器官分化。值得注意的是,两个花烛属物种同时保留了这两个基因,表现出中间的花序性状(具有发达佛焰苞的两性花),这指示了介于祖先和衍生状态之间的过渡进化。这些发现强调了基因保留/丢失如何驱动形态多样化:SOCI的丢失使得吸引传粉者的佛焰苞特化成为可能,而AGL6的丢失促进了繁殖策略的转变。这突显了基因组灵活性作为天南星科花部进化创新的基石。
花烛属佛焰苞的颜色受色素积累的影响,包括花青素、类胡萝卜素和甜菜碱。先前的研究表明,佛焰苞颜色的变异并非仅由花青素决定。例如,矢车菊素-3-芸香糖苷含量及其与天竺葵素-3-芸香糖苷的比率与佛焰苞的亮度(L)和色度参数a相关。与先前发现一致,研究者发现花青素的类型与花烛属的颜色变异没有直接联系。然而,相关性网络显示颜色参数(a、b、C*)与代谢物比率密切相关,表明佛焰苞颜色源于代谢物的协调积累,而非单一色素。虽然花青素仍然是着色的主要贡献者,但类黄酮和类胡萝卜素可能作为颜色强度和色调的调节剂。还鉴定出与花青素和类胡萝卜素生物合成相关的候选基因,突显了它们在佛焰苞着色中的潜在调控功能。然而,重要的是要注意这些关联是相关性的,并不一定表示因果关系。需要进一步的功能验证——如基因编辑或代谢谱分析——来确认这些代谢物和基因在佛焰苞着色中的直接作用。本研究整合分析强调了花烛属花部着色背后复杂的遗传和代谢框架。
植物角质层由角质和蜡质组成,是抵抗环境胁迫和水分流失的关键屏障。在花烛属中,角质层蜡质含量与瓶插寿命相关。化学分析表明,佛焰苞蜡质主要由烷烃、脂肪酸及其衍生物组成,不同品种在发育过程中的组成存在显著差异。特定的烷烃和烷醇可以显著影响蜡质形成。角质层蜡质生物合成涉及通过β-酮脂酰辅酶A合酶、还原酶、脱水酶和烯酰辅酶A还原酶等酶将C16/C18脂酰辅酶A延伸为超长链脂肪酸,随后进入烷烃或醇形成途径。在本研究中,研究者发现CER3和KCS1在“Alabama”和“Xavia”中表现出相似的表达水平。KCS3在“Alabama”和“Xavia”中的差异表达提示了它在蜡质变异中的调控作用。然而,蜡质生物合成的复杂调控需要进一步研究来验证这些发现。
尽管鉴定了如CER3、KCS1、SOCI和AGL6等与角质层蜡质形成和花发育相关的关键候选基因,但它们的功能角色并未进行实验验证,这是当前研究的一个局限。由于缺乏花烛属中高效稳定的遗传转化系统,无法进行基因敲除、过表达或其他功能测定。未来的研究应优先开发适用于花烛属的遗传工具,以便对这些基因进行详细的功能表征。替代方法,如病毒诱导的基因沉默(VIGS)、基于CRISPR/Cas的基因编辑或异源表达系统,也可能为功能验证提供可行的策略。确认这些候选基因的作用对于推进对花烛属佛焰苞蜡质沉积和花发育分子机制的理解至关重要。

END


获取此文献原文PDF、申请加入学术群,联系您所添加的任一微科盟组学老师即可,如未添加过微科盟组学老师,请联系多组学老师30,无需重复添加。
了解更多转录组知识,点击进入《转录组》
点击阅读原文,免费下载该SCI原文

