尽管在通过抑制LOM路径来增强RuO₂基电催化剂在酸性析氧反应(OER)中的稳定性方面已取得了重大进展,但真正参与晶格氧介导(LOM)路径的氧位点仍不明确。
2026年03月26日,深圳大学何传新、胡琪、张雪团队在Advanced Functional Materials期刊发表题为“Enhancing the Stability of Ruthenium Dioxide for Acidic Oxygen Evolution Reaction via Trapping Two-Coordinated Oxygen”的研究论文,团队成员商春彦、亓帅为论文共同第一作者,何传新、胡琪、张雪为论文共同通讯作者。
第一作者:商春彦、亓帅
通讯作者:何传新、胡琪、张雪
通讯单位:深圳大学
论文DOI:10.1002/adfm.75013
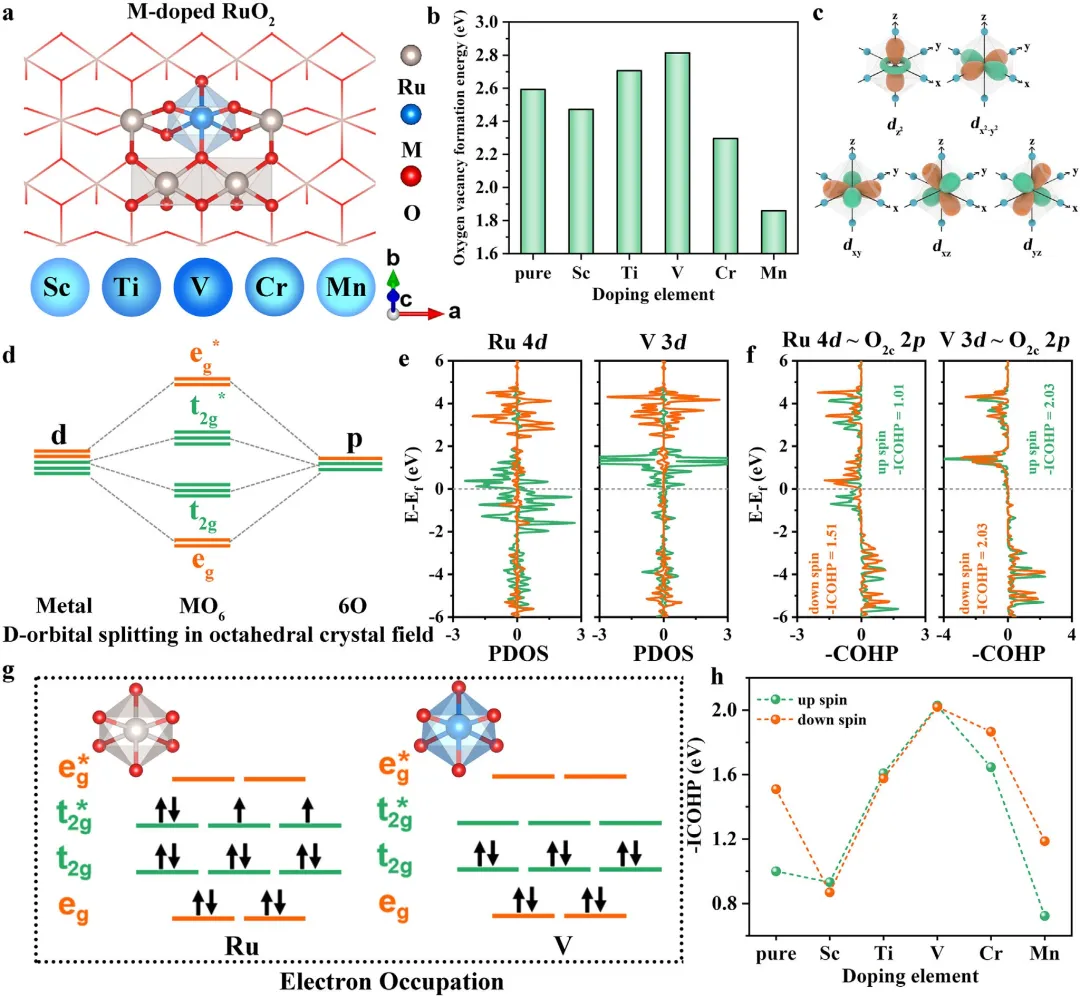
该研究通过系统的密度泛函理论DFT计算发现,RuO₂表面双配位氧(O₂c)位点(即具有较低空位形成能)容易从晶格中释放并参与LOM路径,而三配位氧(O₃c)位点(即具有较高空位形成能)在动力学上对LOM路径是惰性的。为此,研究人员尝试通过引入不对称Ru-O-M共价键构型(M代表Sc、Cr、V、Ti、Mn等金属掺杂剂)来提高O₂c的空位形成能,从而抑制LOM路径。研究发现,与其他构型相比,Ru-O-V构型表现出更高的O₂c空位形成能,因此V掺杂RuO₂能显著抑制LOM路径,从而提高酸性OER的稳定性。上述发现通过合理稳定表面O₂c位点,为提高RuO₂基电催化剂在酸性OER中的长期稳定性开辟了新途径。
质子交换膜水电解槽(PEMWE)被公认为最有效的绿氢生产技术之一,其优势包括高电流密度、优异能效、高纯度氢气输出以及快速动态响应。然而,阳极催化剂稳定性差仍然是阻碍PEMWE系统广泛商业化的巨大障碍。在与PEMWE兼容的催化剂中,二氧化铱(IrO₂)是为数不多的商业化可行选项之一,其在酸性条件下具有强稳定性。然而,Ir是一种稀有贵金属,储量有限且成本高昂,这严重限制了Ir基催化剂的大规模部署。相比之下,Ru基材料(例如RuO₂)在酸性介质中表现出优异的析氧反应(OER)活性,且与Ir相比具有相对更高的自然丰度和更低的成本,使其成为Ir基催化剂有前景的替代品。然而,大多数Ru基电催化剂因高电压和酸性环境而在PEMWE中发生严重降解。因此,设计在酸性OER中兼具长期稳定性和高活性以满足PEMWE要求的Ru基催化剂仍是一项重大挑战。
先前报道的研究已揭示其降解机制,即RuO₂催化剂倾向于遵循晶格氧机制(LOM),并在OER过程中产生大量氧缺陷,导致晶体结构崩塌。为有效抑制Ru基催化剂反应过程中晶格氧的过度参与,并保持其结构与催化性能的稳定性,研究人员提出了一系列有前景的策略,包括缺陷工程、应变效应、界面工程、合金化效应以及掺杂效应。与商业RuO₂相比,这些策略实际上能提升其在酸性条件下的活性和稳定性。其中,客体金属掺杂是一种简便直接的策略,可用于抑制RuO₂的过氧化、稳定晶格氧、调控电子结构与Ru-O共价性,并优化OER过程中中间体的吸附。然而,这些策略主要侧重于通过规避LOM路径来提高RuO₂的酸性OER稳定性,但未能识别出真正参与LOM过程的具体晶格氧位点,也未能实现对这类位点的定向稳定。
在此,该研究首先采用密度泛函理论DFT计算,通过比较表面双配位氧(O₂c)和三配位氧(O₃c)位点的空位形成能,来阐明RuO₂中参与LOM路径的氧位点类型。结果表明,O₃c的空位形成能为3.44 eV,高于O₂c的2.59 eV,表明O₂c是RuO₂在酸性条件下参与LOM反应的主要活性物种。为获得高度稳定的OER性能,研究人员尝试通过引入不对称Ru-O-M共价键构型(M代表Sc、Cr、V、Ti、Mn等金属掺杂剂)来提高O₂c的空位形成能,从而抑制LOM。性能最优的电催化剂V-RuO₂在10 mA cm⁻²下表现出超过1000小时的稳定性测试,其时长是RuO₂的50倍。该研究提出了与RuO₂稳定性相关的新描述符,为后续开发高稳定性酸性OER催化剂提供了新的研究方向。
图1 | (a) M-RuO₂(M = Sc, Ti, V, Cr, Mn)的几何结构。(b) M-RuO₂中O₂c的氧空位形成能。(c, d) 八面体晶体场中d轨道分裂示意图。(e) RuO₂和V-RuO₂中Ru和V的PDOS图。(f) RuO₂和V-RuO₂中Ru-O₂c和V-O₂c的-COHP图。(g) RuO₂和V-RuO₂中Ru和V的电子占据示意图。(h) RuO₂中Ru-O₂c以及M-RuO₂中M-O₂c的-ICOHP值。
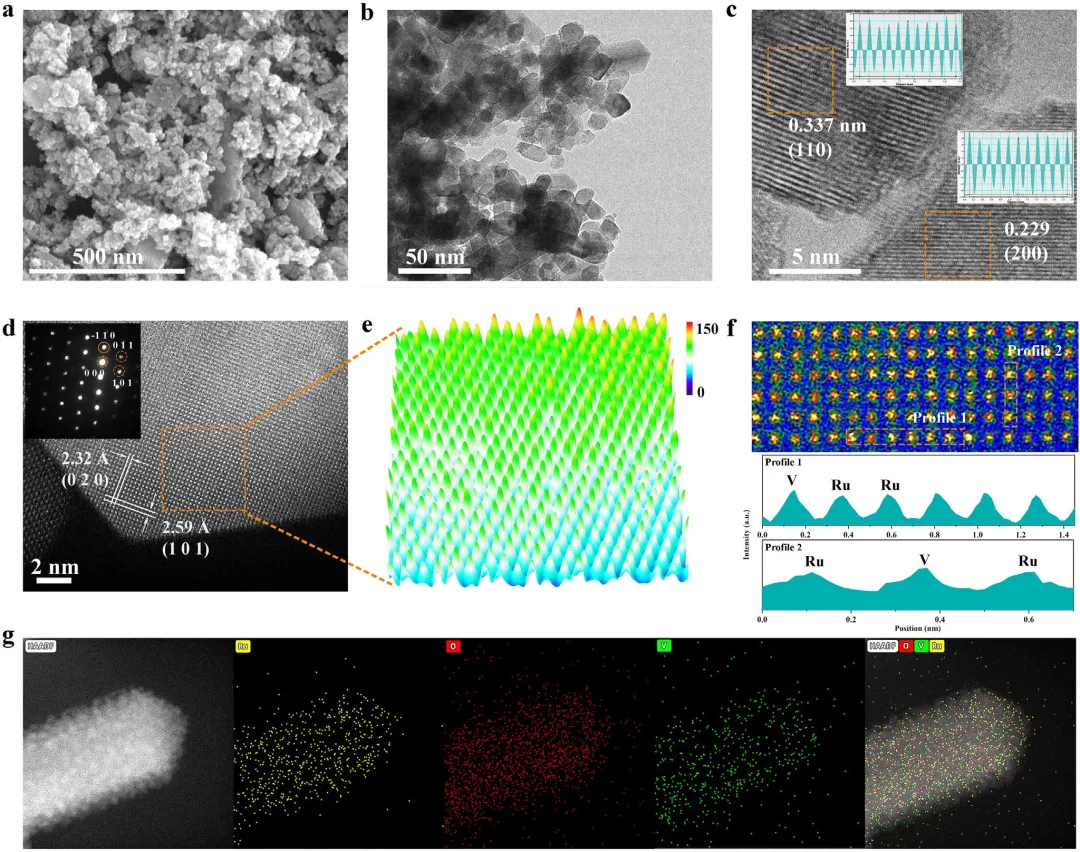
图2 | (a) V-RuO₂的SEM图像和(b)TEM图像。(c) V-RuO₂的高分辨TEM图像。(d) V-RuO₂的HAADF-STEM图像。(e) (d)中选定区域的3D原子重叠高斯函数拟合映射图。(f) (d)中放大区域及对应编号1和2的线扫描轮廓。(g) V-RuO₂的能谱图,显示Ru(黄色)、O(红色)和V(绿色)的均匀分布。
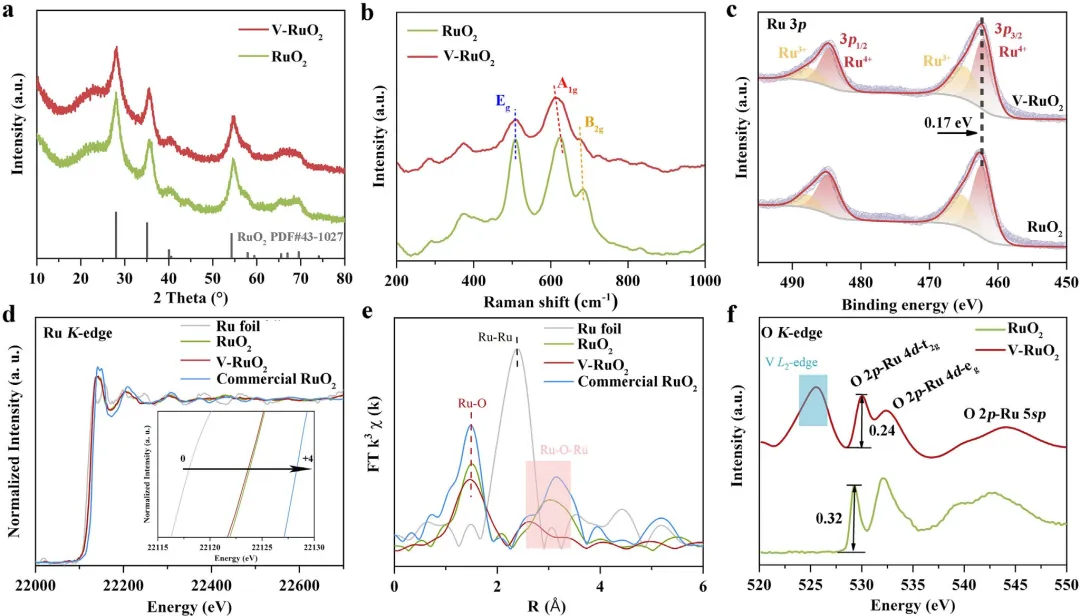
图3 | RuO₂和V-RuO₂的XRD图(a)和拉曼光谱(b)。(c) RuO₂和V-RuO₂的Ru 3p XPS谱图。(d) RuO₂和V-RuO₂在Ru K边的归一化XANES谱图和(e)EXAFS谱图。(f) RuO₂和V-RuO₂的O K边软XAS谱图。
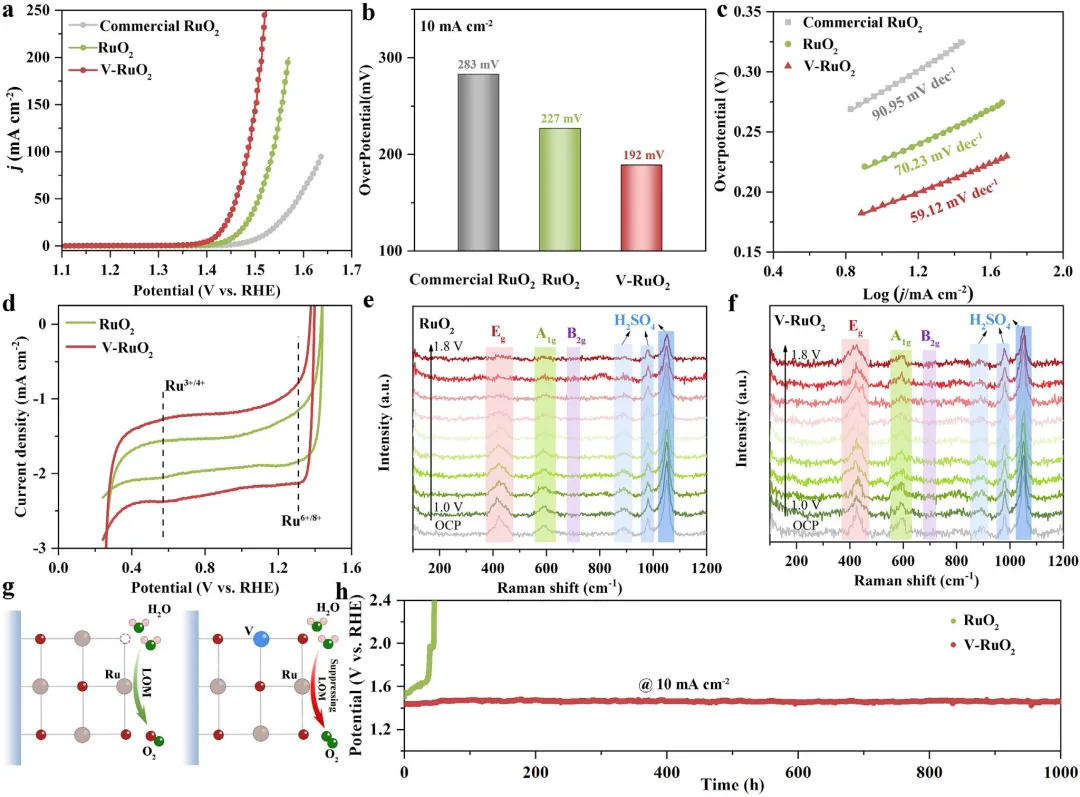
图4 | 商业RuO₂、RuO₂和V-RuO₂的LSV曲线(经iR校正)(a)、在10 mA cm⁻²下的过电位(b)及相应的塔菲尔斜率(c)。(b) RuO₂、RuO₂和V-RuO₂在10 mA cm⁻²下的过电位。(d) RuO₂和V-RuO₂在0.28~1.4 V vs RHE范围内测得的CV分析氧化还原峰。(e, f) RuO₂和V-RuO₂在1.0~1.8 V vs RHE范围内测得的原位拉曼光谱。(g) V-RuO₂在酸性OER中高稳定性的示意图。(h) RuO₂和V-RuO₂在10 mA cm⁻²下的计时电位稳定性曲线。
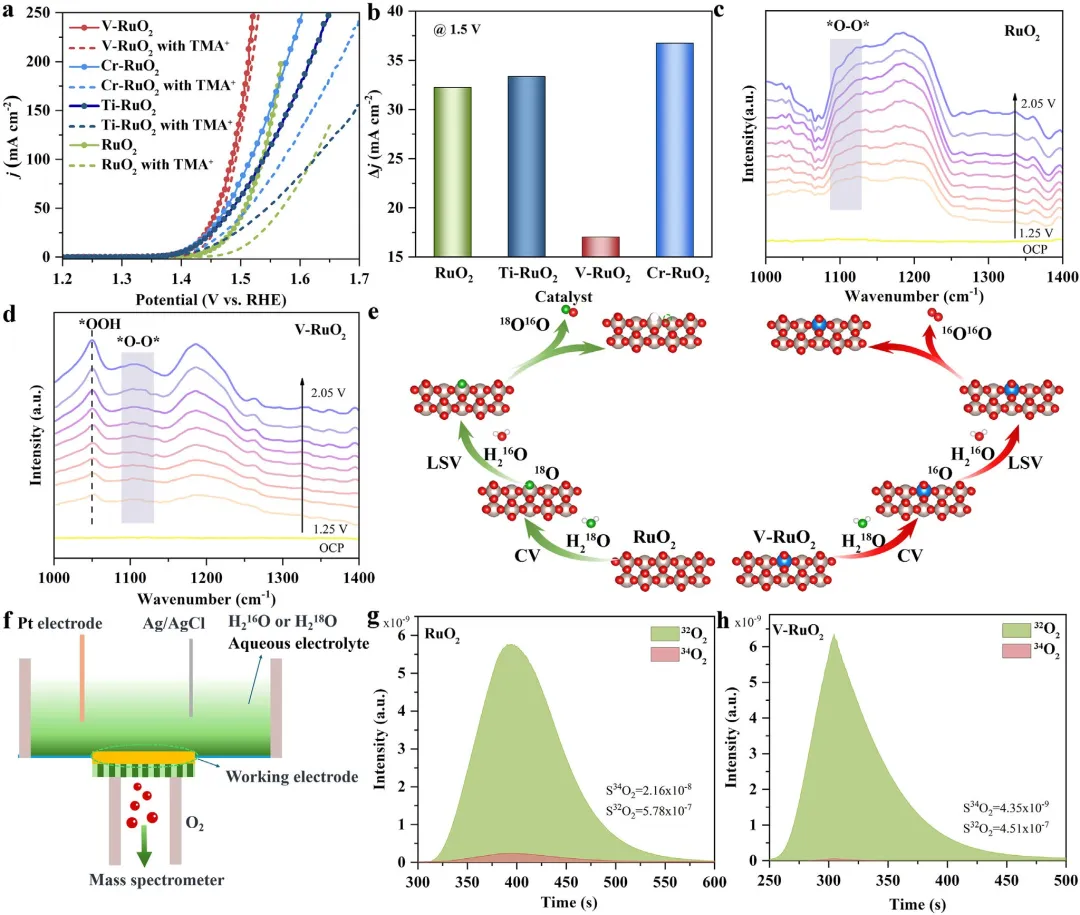
图5 | (a) RuO₂、Cr-RuO₂、Ti-RuO₂和V-RuO₂在添加和不添加TMA⁺时的LSV曲线。(b) RuO₂、Cr-RuO₂、Ti-RuO₂和V-RuO₂在1.5 V vs RHE下添加和不添加TMA⁺的电流密度差值。(c, d) RuO₂和V-RuO₂在1.25~2.05 V vs RHE范围内测得的原位ATR-FTIR谱图。(e) RuO₂和V-RuO₂涉及AEM和LOM路径的DEMS测量示意图。(f) 原位DEMS示意图。(g) ¹⁸O标记的RuO₂和(h) V-RuO₂催化剂在含H₂¹⁶O的0.5 M H₂SO₄电解液中产生氧气的³⁴O₂(¹⁶O¹⁸O)和³²O₂(¹⁶O¹⁶O)的DEMS信号。
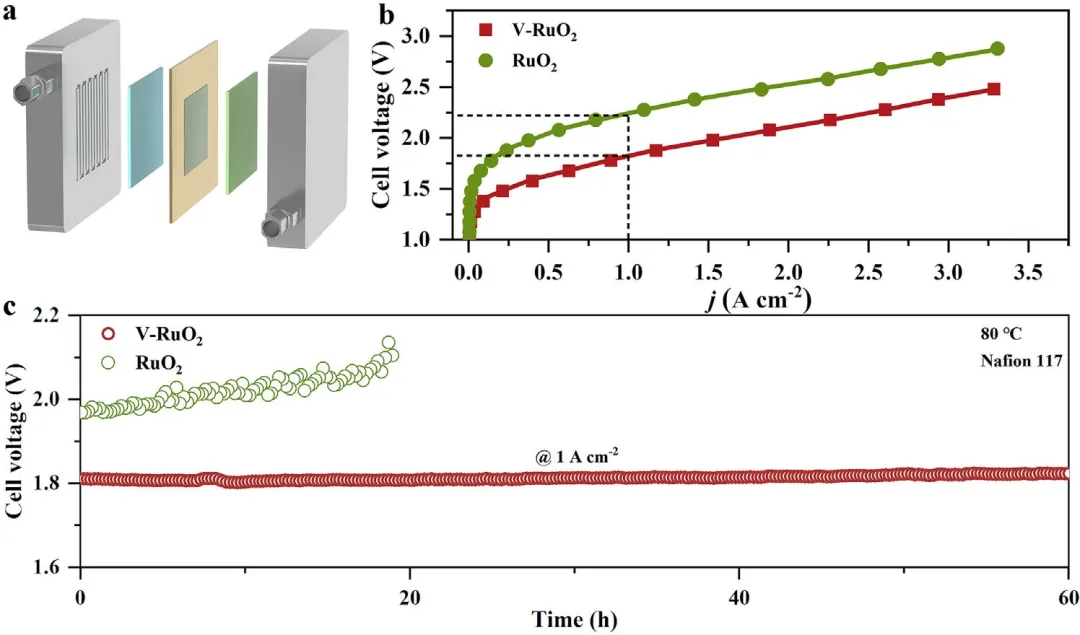
图6 | (a) PEMWE装置示意图。(b) 使用RuO₂或V-RuO₂催化剂作为阳极催化剂、商业Pt/C作为阴极催化剂的PEMWE装置的极化曲线和(c)计时电位曲线。
总之,该研究以RuO₂为模型体系,并利用详细的密度泛函理论DFT计算,系统研究了两种典型表面晶格氧位点(双配位O₂c和三配位O₃c)的空位形成能。计算结果明确证实,O₂c位点是酸性OER中驱动LOM路径的主要活性物种。为验证针对氧位点进行催化剂优化的可行性,进一步构建了一系列具有Ru-O-M不对称共价键构型的M掺杂RuO₂(M = Sc、Cr、V、Ti、Mn),并系统探讨了Ru-O-M不对称共价键网络与氧空位形成能之间的关联。在所有设计的体系中,V-RuO₂表现出最高的O₂c空位形成能,这表明其参与LOM路径的倾向最弱。重要的是,这些计算预测得到了实验结果的充分支持,验证了理论分析和筛选策略的可靠性。最优催化剂V-RuO₂表现出最佳稳定性(在10 mA cm⁻²下超过1000小时),比原始RuO₂提高了50倍。这一发现填补了关于晶格氧具体活性位点的关键理论空白,为调控LOM路径和提高催化剂稳定性提供了精确目标。



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
