恒电势分子动力学新框架:让电化学模拟更贴近真实反应条件
导语:在电化学界面研究中,恒电势条件才是反应的真实写照。然而,受限于传统密度泛函理论代码的整数电子约束,大部分分子动力学模拟仍在“恒电荷”近似下进行。近日,来自中国科学技术大学、深圳先进技术学院和苏黎世大学的研究团队在Journal of Chemical Theory and Computation上发表了一项重要工作——他们在著名的i-PI驱动框架中实现了一套灵活、通用的恒电势模拟方案,成功绕开了整数电子约束,并展现出优异的可移植性和扩展性。让我们一探究竟。
电化学能量转换是未来可再生能源存储与利用的核心技术。想要理性设计电催化剂、理解界面结构与反应路径,必须从原子尺度揭示电化学界面的动态行为。基于密度泛函理论的从头算分子动力学(AIMD)结合显式溶剂和电解质,能够直接捕捉化学键的断裂与形成,已成为研究微观机理的关键工具。
然而,一个根本性的挑战长期存在:真实电化学反应是在恒电极电势下进行的,而绝大多数AIMD模拟仍然是恒电荷的。虽然可以通过添加补偿背景电荷或额外离子在一定程度上“调”电势,但反应前后电势往往发生漂移,难以与实验的恒电势条件直接对应。
从统计力学角度看,恒电势意味着电子化学势和电子数都会在温度下涨落——这正是巨正则系综的采样问题。最近的研究表明,明确考虑微观电势涨落会显著改变预测的反应速率。因此,发展可靠、高效的恒电势AIMD方法,对计算电化学至关重要。
近年来涌现出多种恒电势方法,如Lozovoi等人的可变电子数自洽场、Bonnet等人的扩展哈密顿恒电势器、Bouzid等人的恒费米能级方法、Neugebauer等人的Ne对电极策略等。但多数实现与特定电子结构代码或数值架构深度绑定,跨平台移植性差,也难以与正在兴起的恒电势机器学习势无缝衔接。
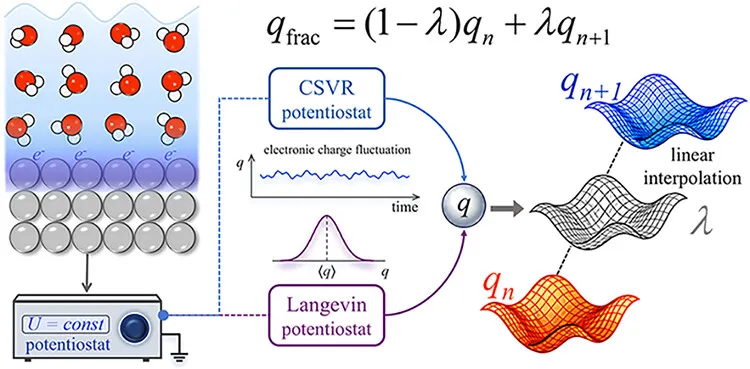
本文的核心创新在于:在i-PI框架内实现了一套模块化、与DFT引擎解耦的恒电势方案,并通过混合哈密顿线性插值技巧,优雅地绕开了CP2K等代码的整数电子约束。同时,作者还实现了两种随机恒电势器——Langevin型和CSVR型,以改善电子自由度的遍历采样。
1️⃣ 绕过整数电子约束的妙招
对于CP2K这类只能使用整数电子的DFT代码,作者采用如下线性插值近似:
· 同时运行两个相邻整数电荷的客户端(如q1和q2)
· 对能量、原子力、功函数(或费米能级)进行线性插值,得到有效“分数电荷”下的物理量:
该方法无需修改底层DFT代码,即可实现连续的电子数响应。
2️⃣ i-PI中新增的恒电势器模块
在Bonnet扩展哈密顿框架基础上,作者为i-PI显式引入了一个电子自由度q及其共轭动量,并设计了专门的恒电势器积分器。为了避免纯确定性热浴的遍历性不足,实现了Langevin和CSVR两种随机恒电势器,并采用BAOAB算子分裂方案确保数值稳定性。
关键参数:电子虚质量 和特征频率 的选取参考了Bonnet的建议,一般使电子模式弛豫时间与核振动谱相近。
3️⃣ 可选Ne对电极方案
受Surendralal等人启发,作者还集成了一个Ne计算对电极选项:通过调整Ne原子上的电子数和核电荷,在保持体系电中性的同时驱动电子转移。这为不依赖隐式溶剂模型的恒电势模拟提供了一条简洁高效的替代路径。
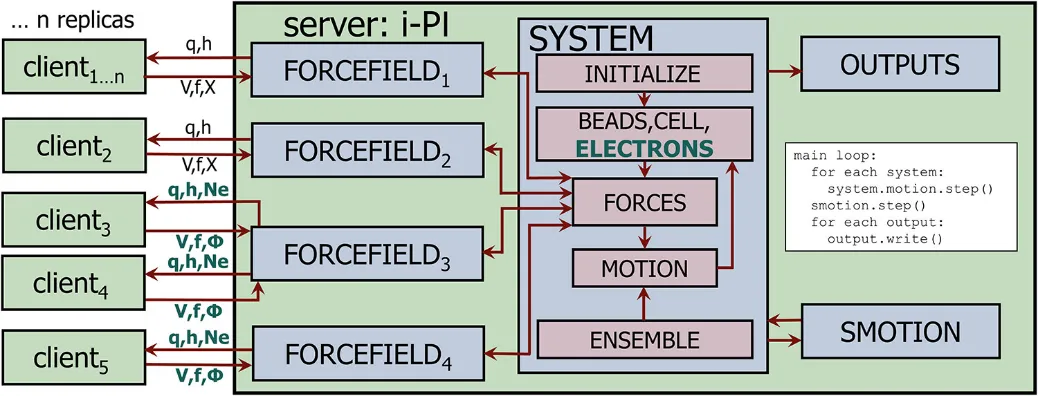
下图展示了修改后的i-PI代码结构与服务-客户端通信协议(图1):
图1:i-PI修改后的代码结构图——新增电子自由度q的通信,恒电势器模块独立控制q的演化,客户端仅需返回整数电荷下的能量、力和费米能级。
✅ 验证一:一维不对称双阱模型
为了验证恒电势器的基本功能,作者构建了一个同时依赖核坐标R和电子电荷q的解析势能面:
图2a展示了该势能面:随净电荷向负方向移动,费米能级相应升高,符合电容模型的预期。
图2b呈现了从不同初始净电荷 出发、采用CSVR和Langevin恒电势器进行的恒费米能级模拟。可以看到:当初始费米能级远离目标值时,体系经历短暂大幅波动后迅速稳定,并围绕目标值平稳涨落——证明恒电势器能够稳健地控制电极电势。
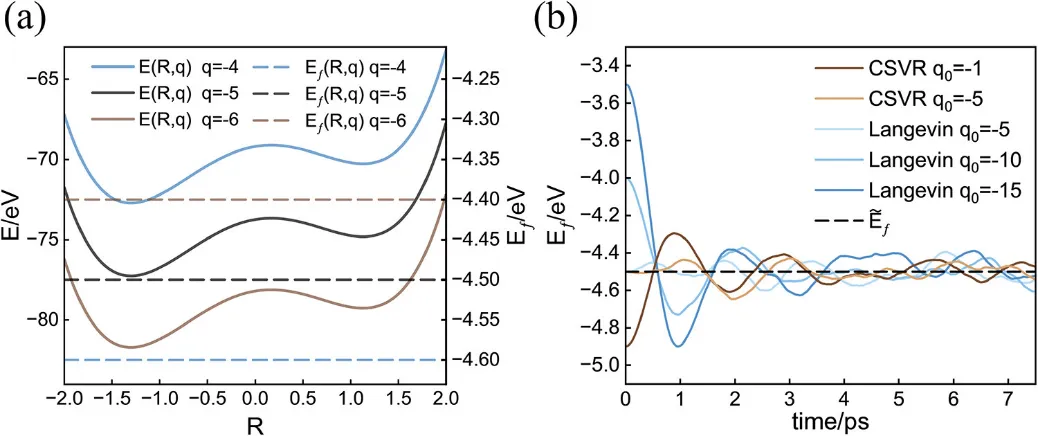
✅ 验证二:Al(111)金属表面
接下来,作者对真实DFT体系——三层Al(111) slab进行了恒费米能级AIMD测试。采用CP2K(PBE泛函、DZVP基组、D3色散修正),通过两个整数电荷客户端(如中性+带负电)线性插值得到连续响应。
图3a显示:当目标费米能级发生阶跃变化时,瞬时费米能级快速响应并进入新的稳态;图3b展示了整个轨迹中电子数分布的直方图——近似高斯分布,符合金属体系巨正则系综的预期。
有趣的是,作者发现将恒电势器温度 设得比核温度 略低,能有效压缩电荷/电势涨落幅度,从而提升SCF收敛性和模拟稳定性——这在实践中非常实用。
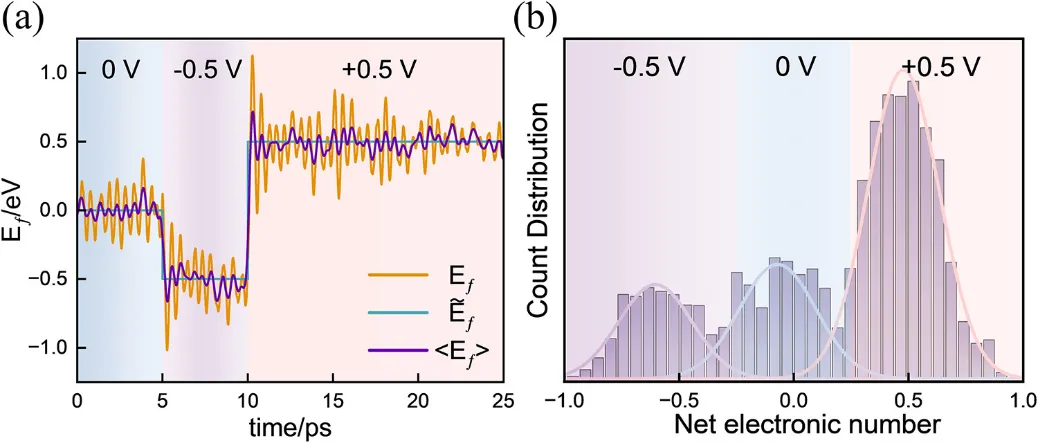
最后的压轴应用是CO₂还原反应(CO₂RR)中关键步 的研究。催化剂为石墨烯负载的Ni单原子(NiN₄配位)。作者分别在 (对应功函数4.1~2.6 eV)下进行恒电势AIMD,采用VASP + 隐式溶剂模型。
图4a展示了模拟体系的结构模型。图4b显示了在-1.0 V下,C–OH键长、净电荷和电极电势随时间的演化——可以看到电势被稳定控制在目标值附近,同时C–OH键涨落反映了反应坐标的探索。
由于在-1.0 V和-1.5 V下未发生自发反应,作者采用well-tempered metadynamics(以C–OH键长为集变量)计算了自由能垒。图4c的结果表明:
· 在-1.0 V下,活化自由能
· 在-1.5 V下,
当电势进一步负移至-2.0 V时,COOH spontaneously 断裂C–OH键生成*CO,甚至观察到甲酸副产物的生成,展示了电势对反应通道的调控作用。值得注意的是,HER反应在模拟时间内未发生,与NiN₄位点对H的高吸附能垒一致。
本文框架的另一重要贡献是为恒电势机器学习势的开发提供了新思路。目前大多数机器学习势在恒电荷数据上训练。本文的混合哈密顿线性插值策略可以这样迁移:
· 在相邻整数电荷端点分别训练两个独立的机器学习模型(每个模型学习该电荷下的能量、力、功函数)
· 模拟时在线性插值两个模型的预测,得到有效分数电荷响应
这避免了直接学习连续电子数依赖所需的大量数据,也无需重新运行昂贵的恒电势AIMD。因此,该框架不仅是一个计算工具,更是一个连接传统DFT与未来机器学习电化学模拟的桥梁。
所有代码和示例输入文件已在GitHub公开:https://github.com/zhangchenyu-USTC/i-pi-constant-potential
作者表示,本文未包含的原始数据可根据合理请求从通讯作者处获取。
本文在i-PI框架中实现了一套灵活、通用、可移植的恒电势分子动力学方案,主要亮点包括:
· ✅ 绕过整数电子约束:混合哈密顿线性插值,无需修改DFT代码
· ✅ 两种随机恒电势器(Langevin / CSVR),改善电子自由度的遍历采样
· ✅ 可选Ne对电极,提供隐式溶剂之外的简易电势控制手段
· ✅ 验证充分:从解析模型到金属表面,再到真实的CO₂RR催化体系
· ✅ 开源实现,易于扩展至其他DFT引擎及未来的机器学习势
这项工作为电化学界面的巨正则系综模拟提供了一个高效、开源的平台,有望推动恒电势AIMD在电催化、腐蚀科学、能源材料等领域的广泛应用。
DOI: 10.1021/acs.jctc.6c00504


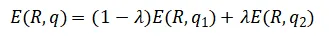
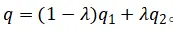
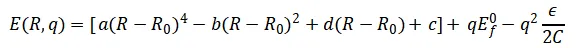
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
