原文标题:Stabilizing High-Activity FeN5 Sites via an Adaptive N-linked Carbon Bilayer for Stable Fuel Cells通讯作者:隋旭磊,王振波
通讯单位:深圳大学、哈尔滨工业大学
论文DOI:10.1002/anie.6494121
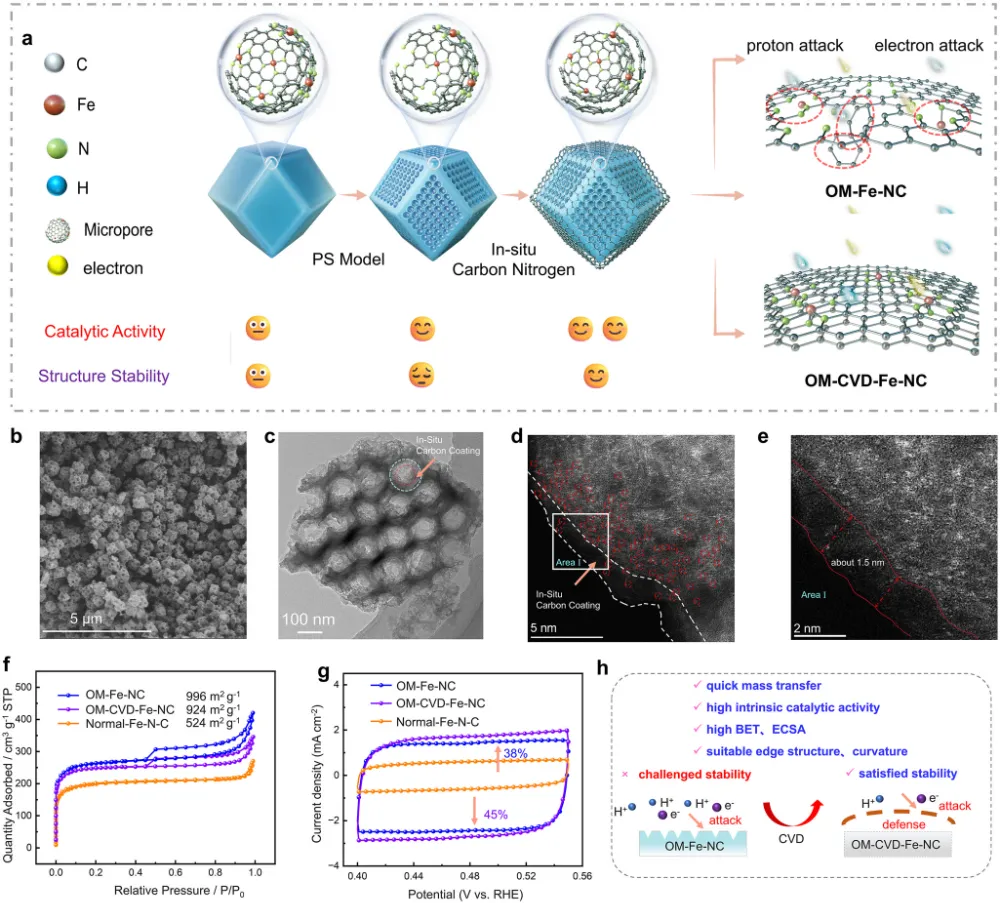
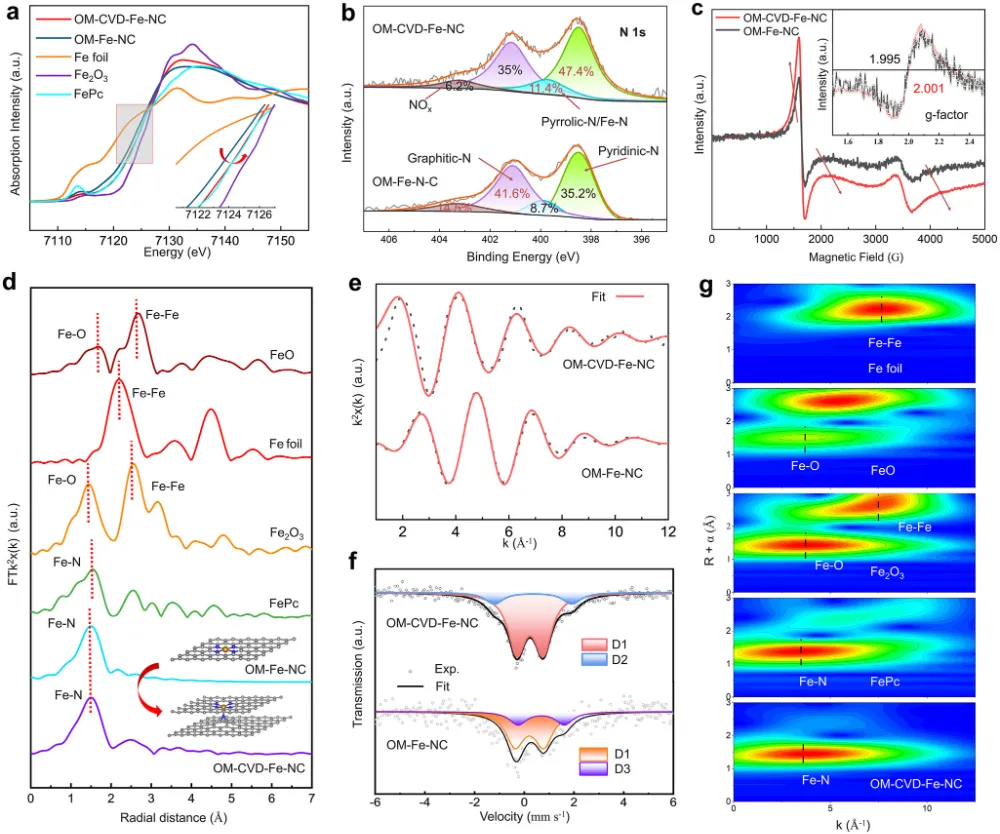
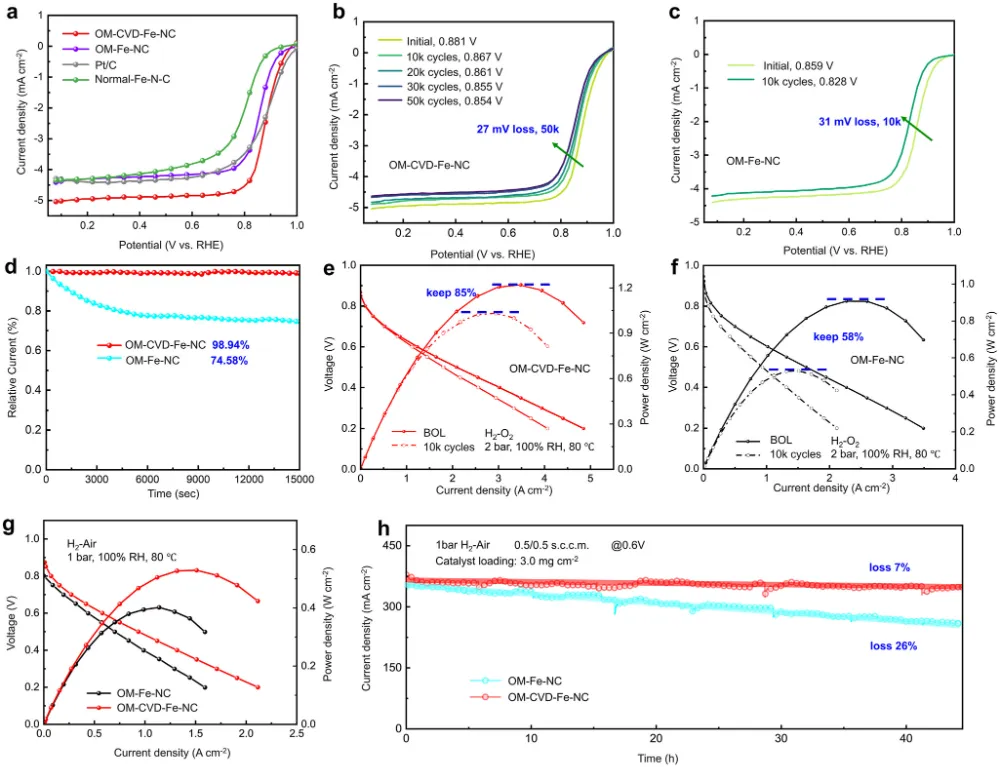
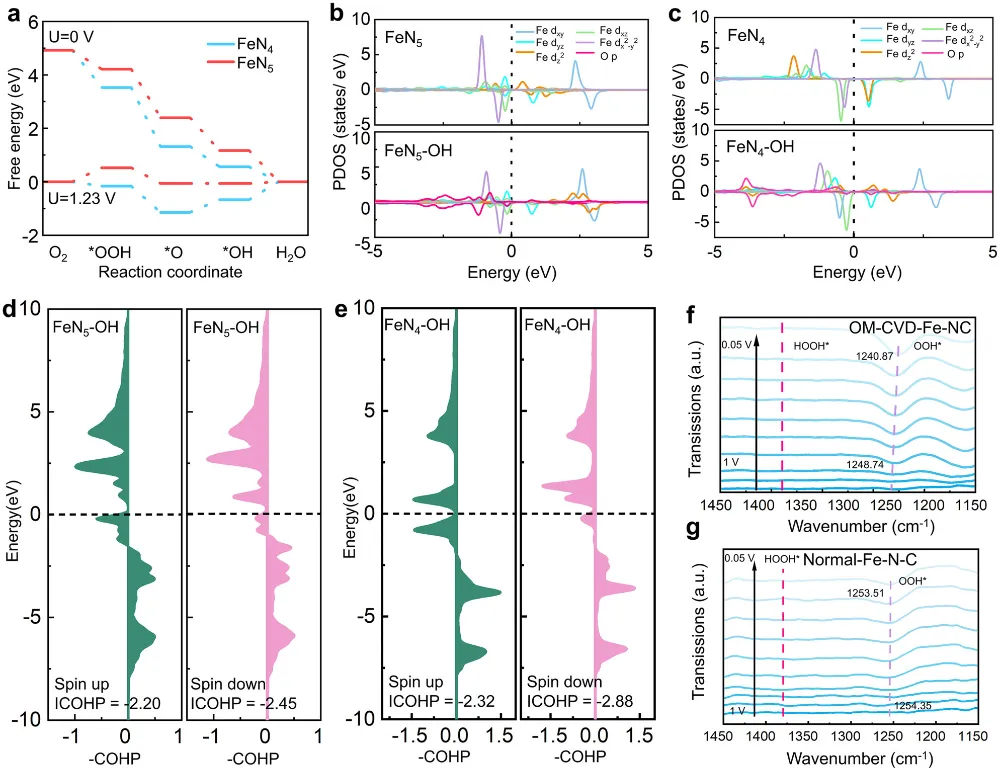
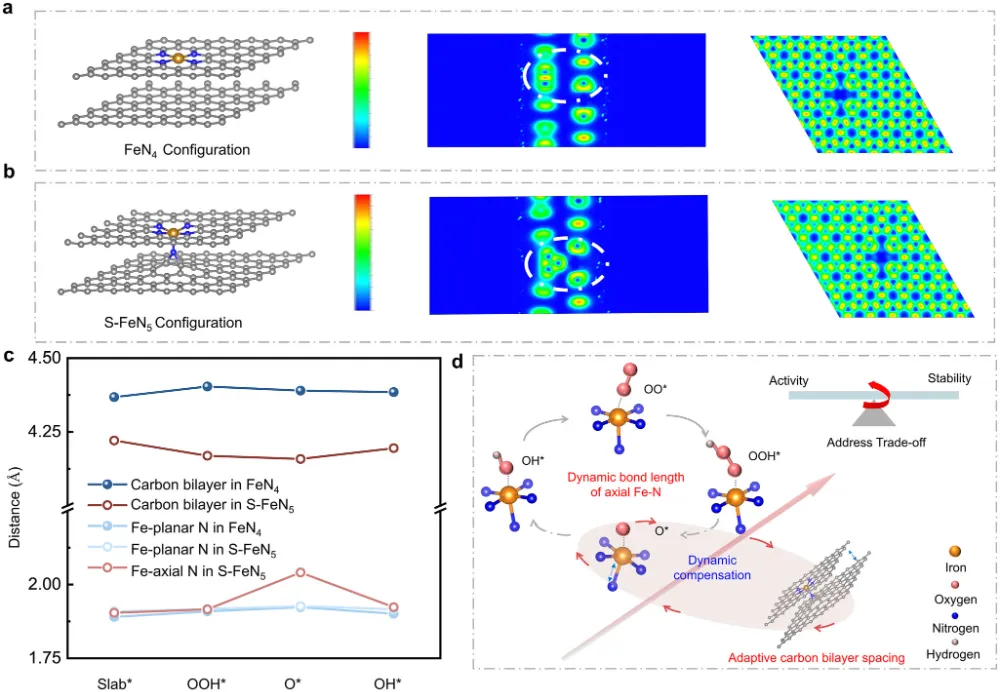
期刊与年份:Angewandte Chemie International Edition,2026文章类型:质子交换膜燃料电池 / 酸性氧还原 / Fe-N-C 单原子催化剂这篇文章围绕 Fe-N-C 单原子催化剂最棘手的老问题展开:在酸性氧还原反应中,活性和稳定性往往很难同时提高。作者没有继续在“多做孔”“多暴露位点”这种常规结构优化层面打转,而是把问题推进到活性位局域环境这一更关键的尺度,提出一种“修复并升级”的工程策略:一边在热解过程中原位补充碳和氮,修补高温热解造成的缺陷;一边把常规 FeN4 位点推进为具有轴向氮配位的 FeN5 位点,并让其被一个 N 键连的碳双层局域包围。全文最有分量的地方在于,作者不仅证明了轴向 N 配位能够提升 Fe 中心电子结构、减弱 *OH 过强吸附,从而提高活性;还进一步提出,这个碳双层并不是静态包覆层,而是能够在反应过程中自适应调节层间距,补偿轴向 Fe-N 键长波动,持续锁定高活性位点。这使文章从“做出一个更好 Fe-N-C 催化剂”上升为“提出一种解决活性-稳定性权衡的局域环境设计原则”。广泛背景:Fe-N-C 单原子催化剂被视为替代铂族金属用于质子交换膜燃料电池阴极氧还原反应的重要路线 → 核心问题:传统 FeN4 位点虽然可能具有较高活性,但在酸性与高电位条件下容易发生脱金属、碳腐蚀和结构失活,活性与稳定性长期相互掣肘 → 现有理解及其局限:多级孔结构与轴向配位工程分别有助于传质或电子调控,但常常彼此割裂,且轴向配位在真实反应中本身也可能不够稳定 → 本文核心观点:通过热解过程中原位 CVD 补充碳氮,可同步修复缺陷、构建 N 键连碳双层,并把 FeN4 升级为稳定的 FeN5 活性位 → 假设的机制链条:轴向 N 改写 Fe 中心电子结构,减弱 *OH 过强吸附;碳双层层间距在 ORR 动态过程中自适应变化,补偿轴向 Fe-N 键长波动,机械性锚定高活性位点 → 关键证据:XAS、EPR、穆斯堡尔谱确认高自旋 Fe(III)-N5 形成;电化学与 MEA 测试显示 E1/2 达 0.881 V、峰值功率密度达 1221 mW cm−2 且 10000 圈后仍保留超过 85% 初始功率;DFT 和 ATR-SEIRAS 证明 *OH 吸附减弱且局域结构动态更稳 → 研究成果与启示:局域配位与周围碳骨架的协同适配,是打破单原子催化剂活性-稳定性矛盾的一条可迁移设计原则。这篇文章真正精彩的地方,在于它并没有把 Fe-N-C 催化剂失活简单理解成“Fe 位点不够稳”或者“碳载体容易腐蚀”这类孤立问题,而是把金属中心、配位配体与周围碳骨架放在同一个动力学框架下讨论。作者提出的金属-配体-基体三元耦合视角,本质上是在问:活性位为何在工作中会逐步丢失其最优构型,而我们能否设计一种局域环境,让它在反应过程中始终被拉回到更稳定、更高效的状态。全文的引爆点是“修复并升级”这个双重操作。以往很多工作会分别做两件事:要么修补碳缺陷、加一层保护碳壳;要么通过额外配位原子去改电子结构。但这篇文章把两者合并进同一个原位 CVD 步骤里,使碳和氮的补充不只是改善外层结构,更直接推动了 FeN4 向 FeN5 的升级,并且留下了一个可随反应自适应呼吸的 N 键连碳双层。从论证结构看,这不是简单的性能堆砌,而是结构表征、谱学、电化学、膜电极验证和理论计算的逐层收束。作者先通过 XAS、EPR 与穆斯堡尔谱把高自旋 Fe(III)-N5 位点“坐实”,再用 RRDE 与 MEA 数据证明其活性和稳定性确实兼得,最后用 DFT 和原位 ATR-SEIRAS 回答为什么它能做到这一点。尤其“层间距自适应补偿 Fe-N 键长波动”这一点,把通常静态理解的碳包覆层转化成了具有动态响应能力的结构单元。文章另一个值得重视的地方,是它没有停留在旋转圆盘电极层面的半反应数据,而是进一步推到质子交换膜燃料电池膜电极组件中,给出了 1221 mW cm−2 的 H2-O2 峰值功率密度、H2-air 条件下 528 mW cm−2,以及 10000 次方波循环后仍保持 85% 初始功率的结果。这使这项工作不仅是机理意义上的改进,也具备相当明确的器件转化指向。如果把这篇文章当成研究训练样本来看,最值得学习的是它的思路不是“单纯找更强的 Fe 位点”,而是围绕高活性位点为什么在真实工况下失真和失活,去设计一个能够动态跟随、动态缓冲的微环境。这种从静态最优结构转向动态稳定结构的思维,对单原子催化剂、双原子催化剂乃至更广泛的电催化界面设计都很有启发。这一部分是全文最核心的技术主轴。作者的关键贡献,并不是仅仅通过原位 CVD 做了一层碳包覆,而是同时实现了结构缺陷修复、Fe 配位环境升级以及多级孔结构保留,从而把活性与稳定性这两条通常相互制约的路径压缩到同一设计中。1. 实验体系与修复升级策略:作者先利用 3D 有序聚苯乙烯模板和 Fe-ZIF-8 前驱体构建有序大孔骨架,再在高温热解时引入原位化学气相沉积,同步补充碳和氮。这样形成的 OM-CVD-Fe-NC 既保留了约 70 nm 的开放大孔结构,又在局域上形成双层碳环境。与未经过 CVD 的 OM-Fe-NC 相比,这一步不是简单增厚碳层,而是在热解后期重新塑造局域化学环境。2. 活性位从 FeN4 升级到 FeN5:XANES、EXAFS 拟合结果给出 Fe-N 配位数约 5.1,说明 OM-CVD-Fe-NC 中 Fe 单原子位点已被五个 N 原子包围;而 OM-Fe-NC 的 Fe-N 配位数约 4.1,更接近传统 FeN4。与此同时,XPS 中吡啶型 N 比例上升、Fe3+ 相对含量提高,EPR 与 57Fe 穆斯堡尔谱进一步证明高自旋 Fe(III) 物种占主导。换句话说,原位 CVD 的作用不是外部附着,而是把活性中心化学本性真正改写了。3. 传质结构与位点可达性并未因保护层而牺牲:尽管 OM-CVD-Fe-NC 比 OM-Fe-NC 略微降低了比表面积,但其电化学活性表面积仍较普通样品显著提升,可达位点密度也达到 3.03 × 10^19 sites g−1。这里的关键在于,作者构建的是内层较致密、外层较疏松的双层碳结构,使保护作用与传质通道可以共存,而不是彼此对冲。4. 电化学活性与耐久性实现同时领先:OM-CVD-Fe-NC 的半波电位达到 0.881 V,0.85 V 下动力学电流密度为 14.70 mA cm−2,质量活性达到 9.10 A g−1,TOF 达 11.20 e site−1 s−1,均显著优于对照样。更重要的是,其在 10000 次加速循环后 E1/2 仅损失 14 mV,50000 次内继续衰减也较小;恒电位 15000 s 测试中电流保持率高达 98.94%。这说明文章追求的不是短时活性峰值,而是真正在酸性环境中稳住高活性位。5. 膜电极与器件测试验证工程价值:在 H2-O2、2.0 bar、80 °C 条件下,OM-CVD-Fe-NC 的峰值功率密度达到 1221 mW cm−2,优于 OM-Fe-NC 的 907 mW cm−2;在 H2-air 下仍有 528 mW cm−2。10000 次方波循环后功率仍保留 85%,而 OM-Fe-NC 仅保留 58%。更长时间的 0.6 V H2-air 恒电位测试超过 45 h 几乎无衰减,说明这种局域环境设计已经具有明确的燃料电池应用指向。综合来看,作者通过“轴向电子调控 + 自适应力学补偿 + 有序孔传质”三件事协同发力,提出了一种比单纯加强配位或单纯加保护层更完整的稳定化方案。研究背景:Fe-N-C 催化剂是替代铂族金属用于质子交换膜燃料电池阴极氧还原反应的核心候选,但实际应用长期受制于活性和稳定性之间的内在权衡。存在的挑战/问题:传统 FeN4 位点即便可以表现出较高活性,也常因酸性工况下的自由基攻击、脱金属和碳腐蚀而快速失活;同时,单纯提高孔结构或引入轴向配位,并不能自动解决长期稳定性问题。研究方案:作者提出“修复并升级”工程策略,在热解过程中通过原位 CVD 同步补充碳和氮,一方面修复高温热解造成的结构缺陷,另一方面将 FeN4 位点演化为被 N 键连碳双层局域保护的 FeN5 位点,并用多种表征、电化学测试、膜电极测试和理论计算完成验证。核心亮点与发现:1. 原位 CVD 促成传统 FeN4 向高自旋 Fe(III)-N5 位点升级,并提高吡啶型 N 比例。2. 轴向 N 配位优化 Fe 中心电子结构,削弱 *OH 过强吸附,降低 ORR 过电位。3. N 键连碳双层在反应中可自适应调节层间距,补偿轴向 Fe-N 键长变化,稳定高活性位。4. 催化剂在 RRDE 与 MEA 中同时表现出高活性和高稳定性,峰值功率密度达 1221 mW cm−2,10000 圈后仍保留超过 85% 初始功率。研究意义与展望:摘要最后强调的不是又一个高性能 Fe-N-C 样品,而是一种面向单原子催化剂的普适设计原则,即通过创造可动态适配的局域环境,系统性缓解活性-稳定性权衡。创新点一:把热解后的“修补缺陷”和“升级活性位”整合成同一个原位 CVD 步骤。数据支撑:图1 展示了原位氮碳补充策略和有序大孔结构形成过程;图2 的 XAS 拟合显示配位数由约 4.1 提高到约 5.1,说明 FeN4 被推进为 FeN5。创新点二:提出轴向 N 配位与周围碳双层并非独立设计,而是共同决定活性位表现。数据支撑:图2 的 XPS、EPR 和 57Fe 穆斯堡尔谱表明 OM-CVD-Fe-NC 中高自旋 Fe(III)-N5 占主导,这说明活性中心电子结构与局部基体结构同步被改写。创新点三:用动态自适应碳层解释高活性位为何能长期稳定。数据支撑:图5 的理论结果显示,反应过程中碳双层层间距与轴向 Fe-N 键长呈补偿式变化,从而缓冲结构畸变,稳定 S-FeN5 位点。创新点四:在活性、耐久性和膜电极器件表现三个层面同时证明该设计有效。数据支撑:图3 显示 E1/2 达 0.881 V、10000 圈后仅衰减 14 mV;H2-O2 条件下峰值功率密度达 1221 mW cm−2,10000 次方波循环后仍保留 85% 初始功率。创新点五:提出金属-配体-基体三元协同的新设计视角。数据支撑:图4 和图5 共同表明,轴向配位不仅改变中间体吸附能,还通过与碳基体的协同适配降低了位点在真实反应中的失稳概率。前言开头先从质子交换膜燃料电池对非贵金属阴极催化剂的需求切入,指出 Fe-N-C 单原子催化剂是替代铂族金属的代表性方向。这一层背景并不新,但作者紧接着把矛盾收束得很明确:FeN4 位点的热力学过电位依然不低,而在酸性工况下又容易发生自由基攻击、脱金属与碳支撑腐蚀,因此所谓“高活性位”往往也是最脆弱的位点。第二层推进发生在结构工程层面。作者回顾了多级孔结构设计如何帮助传质,也讨论了轴向配位如何调控 Fe 中心 d 轨道与中间体吸附,但明确指出这两条路线多数情况下是分开的。前者能改善位点可达性,却不能直接保证位点本身稳定;后者虽有机会提升本征活性,但轴向配位键在真实电催化循环中本身也可能断裂。文章最关键的文献缺口定位,在于当前缺少一种既能构造高活性轴向配位位点,又能保证其在酸性 ORR 动态环境中持续稳定的策略。作者因此不满足于“再造一个 FeN5”,而是进一步追问:能否同时在位点周围构建一个能够机械补偿其动态畸变的局域碳环境。前言最后一段实际上给出了全文路线图。作者预告将通过原位 CVD 同步补充碳和氮,既修复热解缺陷又升级 Fe 配位环境,并用结构表征、电化学、膜电极性能和理论计算说明这种 N 键连碳双层如何在反应中保护高活性 FeN5 位点。这样读者从一开始就知道,本文不是单点性能优化,而是试图回答一个关于单原子催化剂为何在工作中失稳、又如何被动态稳住的更深层问题。来源:原文图1,Angew. Chem. Int. Ed.,2026原文题录:FIGURE 1. Stability-enhanced engineering by operando nitrogen-carbon coating synthesis; SEM, TEM, HAADF-STEM, pore structure, and ECSA analyses.中文解读:该图从合成策略、形貌结构、单原子分散状态和孔结构出发,证明作者构建了兼具有序大孔与局域碳双层的 OM-CVD-Fe-NC 催化剂。图文解析:这张图在全文中的作用是把“修复并升级”这个概念落到真实可观察结构上。读图重点包括:有序大孔仍然存在,说明传质通道没有因为保护层而被堵死;HAADF-STEM 中没有 Fe 团簇,说明单原子分散得以保留;ECSA 和位点密度结果又表明原位 CVD 并未牺牲位点可达性。换句话说,作者不是用一层厚重保护层换稳定,而是在保留开放结构的同时重塑了局域环境。读者应带走的结论:这一步先把“结构可行”坐实,后面的活性与稳定性提升才有可信基础。图2. FeN5 位点的形成与高自旋 Fe(III) 电子结构确认来源:原文图2,Angew. Chem. Int. Ed.,2026原文题录:FIGURE 2. Fe K-edge XANES, N 1s XPS, EPR, EXAFS fitting, 57Fe Mössbauer spectra, and WT-EXAFS of OM-CVD-Fe-NC and controls.中文解读:该图综合 XPS、XANES、EXAFS、EPR 和穆斯堡尔谱,核心任务是证明 OM-CVD-Fe-NC 中活性位已经由传统 FeN4 升级为高自旋 Fe(III)-N5。图文解析:对全文而言,这张图是机制链的起点。XPS 显示吡啶型 N 和 Fe3+ 比例上升,说明原位氮补充确实改变了配位环境;EXAFS 拟合给出约 5.1 的 Fe-N 配位数,是从结构层面最直接的证据;EPR 与穆斯堡尔谱进一步把这种位点锁定为高自旋 Fe(III) 主导。读者应注意,作者真正强调的不是‘多了一个 N’这么简单,而是 Fe 中心电子结构和周围碳氮骨架一起被重新组织。读者应带走的结论:FeN5 的形成是全文所有性能提升与稳定性解释的中心前提。来源:原文图3,Angew. Chem. Int. Ed.,2026原文题录:FIGURE 3. ORR polarization curves, accelerated durability tests, chronoamperometry, MEA polarization/power density curves, and long-term H2-air operation.中文解读:该图集中展示 RRDE 半电池性能、加速老化结果以及膜电极组件中的功率和长期运行表现,是全文最直接的性能证据。图文解析:最核心的信息有三层。第一,OM-CVD-Fe-NC 的半波电位达到 0.881 V,动力学电流、质量活性和 TOF 全面领先,说明 FeN5 位点确实更活跃。第二,10000 次循环后 E1/2 仅损失 14 mV,恒电位保持率接近 99%,说明改进不是短时峰值效应。第三,进入 MEA 后峰值功率密度达到 1221 mW cm−2,并在 10000 次方波循环后仍保留 85% 初始功率,这说明局域环境设计能真正穿透到器件层面。读者应带走的结论:这张图把作者的主张从‘结构更优’推进到‘应用上也更可靠’。图4. 轴向 N 如何减弱 *OH 吸附并重写 ORR 速控步骤来源:原文图4,Angew. Chem. Int. Ed.,2026原文题录:FIGURE 4. ORR free-energy diagrams, PDOS, COHP/ICOHP analyses, and in situ ATR-SEIRAS for FeN4 and S-FeN5.中文解读:该图给出自由能图、电子态密度、成键强度分析和原位红外证据,用来解释 FeN5 为什么比 FeN4 活性更高。图文解析:这里最重要的不是一句‘FeN5 更好’,而是作者把好在哪里拆开了。自由能图显示 S-FeN5 的 ORR 过电位更低,说明速控瓶颈被缓解;PDOS 与 COHP 结果说明轴向 N 让 Fe 3d 与 O 2p 的相互作用位置更接近费米能级,从而削弱 *OH 过强吸附;原位 ATR-SEIRAS 则从实验上看到氧中间体吸附行为的差异。也就是说,活性提升来自电子结构重新分配,而不是简单增加位点数量。读者应带走的结论:FeN5 的价值在于把传统 FeN4 上过强的 *OH 束缚拉回到更合适的窗口。图5. 自适应碳双层如何在反应过程中稳定高活性 FeN5来源:原文图5,Angew. Chem. Int. Ed.,2026原文题录:FIGURE 5. Optimized structures, electron localization function of FeN4 and S-FeN5, adaptive carbon-bilayer spacing, and schematic of resolving the activity-stability trade-off.中文解读:该图是全文最具方法论意义的部分,它把自适应碳双层从概念图推进为可计算、可解释的稳定化机制。图文解析:读图时要抓住两个核心关系。第一,轴向 N 把 Fe 中心从平面 FeN4 中拉出,增强电子离域并提高构型稳定性。第二,更关键的是碳双层层间距会随反应过程中轴向 Fe-N 键长变化而反向调节,像一个局部缓冲器那样持续补偿结构应变。作者因此把单原子位点稳定性从‘某一时刻结构够不够稳’提升为‘反应循环中能否持续自我调节’这一动态问题。读者应带走的结论:这张图给出的不是单一材料细节,而是一种可迁移的稳定化设计框架。从整体科学贡献看,本文最重要的突破并不是又一次把 Fe-N-C 催化剂做得更活跃,而是提出了一个更完整的稳定化思想:真正决定单原子催化剂长期表现的,不只是金属中心和第一配位层,还包括围绕其工作的可动态适配的碳基体。作者把这一点概括成金属-配体-基体三元协同,逻辑上是成立且有证据支撑的。从机理深度看,FeN5 的优势来自两个层面同时发生。一个是电子层面,轴向 N 把 Fe 中心从 FeN4 的过强 *OH 吸附状态中拉出来,使 ORR 关键中间体结合更接近最优窗口;另一个是力学与构型层面,N 键连碳双层在反应中可以持续补偿 Fe-N 键长波动,避免高活性位在循环中逐渐走向失真和失活。正是这两个层面的叠加,才让高活性与高稳定性不再非此即彼。从方法学角度看,这篇文章很值得借鉴的地方在于,它没有把碳包覆当成单纯防护层,也没有把轴向配位当成单纯调电子结构的手段,而是把两者设计成同一个动态体系。今后在单原子催化、双原子催化甚至酶模拟位点设计中,这种‘让周围基体跟着活性位一起工作’的思想都很有推广潜力。当然,文章仍有一些值得继续追问的边界。首先,当前结论主要建立在 Fe-N-C 与 PEMFC 阴极 ORR 体系中,这种自适应碳双层是否可无损迁移到其他过渡金属中心或不同反应环境,还需要更多验证。其次,文中虽然通过理论和原位谱学提供了有力支持,但对极长时间运行后局域双层是否仍保持同样响应能力,仍值得更长时程原位追踪。未来最值得推进的方向,可能包括三类:一是把局域碳层的自适应能力抽象为可计算描述符,用于前置筛选稳定高活性位点;二是把该策略推广到双原子或异核位点体系,观察是否存在更复杂的协同补偿;三是在真实燃料电池工况下结合更多原位结构手段,直接追踪碳双层与 Fe-N 键的协同演化。如果这些问题继续向前推进,本文提出的设计原则很可能成为单原子燃料电池催化剂研究中的一个长期框架。论文来源:Yuan LJ, Miao ZY, Li Q, Liu YZ, Gong T, Han ZY, Gong W, Shen LX, Feng WL, Wu B, Yuan SR, Zhang GX, Sui XL, Wang ZB. Stabilizing High-Activity FeN5 Sites via an Adaptive N-linked Carbon Bilayer for Stable Fuel Cells. Angewandte Chemie International Edition. 2026. DOI: 10.1002/anie.6494121。课题组 / 作者介绍
隋旭磊:深圳大学副教授/特聘研究员,博士生导师。本硕博均毕业于哈尔滨工业大学(导师王振波教授),加拿大西安大略大学博士后(导师孙学良院士)。研究方向为新型化学电源及纳米电催化,主要从事质子交换膜燃料电池催化剂、膜电极和锂钠离子电池电极材料的设计与制备。近年来,主持国家自然科学基金面上项目、青年项目,中国博士后科学基金特别资助等10余项基金。已在Nat. Commun.、Adv. Mater.、 Angew. Chem. Int. Ed.、Adv. Energy Mater.、Adv. Funct. Mater. 等发表SCI文章100余篇,H因子41,获省自然科学一等奖1项。
王振波:哈尔滨工业大学二级长聘教授,博士生导师。入选国家级高层次人才(第四批)、科技部中青年科技创新领军人才;黑龙江省“龙江学者”特聘教授;获2022年侯德榜化工科学技术奖创新奖;入选2025年科睿唯安“全球高被引科学家”;连续12年入选Elsevier中国高被引科学家。研究方向为先进化学电源、氢燃料电池、电催化、纳米电极材料;主持国家自然科学基金重点基金1项,面上项目3项,青年基金1项;科技部重点研发项目1项、课题1项;山东省重点研发项目1项,其他省部委项目10项,其他及企业课题40多项。在Nature Catalysis、Nature Commun.、Adv. Mater.、Angew. Chem. Int. Ed.等上发表SCI论文350多篇,H因子75。获国家授权发明专利68项,转化43项;获国防科技进步一等奖1项,黑龙江省自然科学一等奖2项。
博士生及博士后招收:课题组长期招收博士生和博士后,欢迎具有质子交换膜燃料电池催化剂与膜电极、锂钠离子电池等先进化学电源相关研究背景或兴趣的博士生申请。

 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
