江南大学娄阳&中科院深圳先进院郑勇平&华东理工大学詹望成最新Nature子刊丨1T′-MoS₂空间耦合活性位点实现近100%甲醇选择性!
- 2026-07-16 15:49:45

甲醇作为重要的液态能源载体和化工原料,其绿色合成对能源转型具有重要意义。将CO₂选择性加氢转化为甲醇是缓解温室效应和能源危机的可持续策略,但受限于CO₂的热力学稳定性(C=O键能803 kJ/mol)和反应中间体精准调控的困难。
关键中间体*CO的C-O键断裂与否直接决定产物是甲醇还是甲烷,因此理想催化剂需同时实现C=O键高效活化和C-O键稳定保持。受自然界酶催化多中心协同机制的启发,串联催化系统通过空间邻近的活性位点协作驱动多步反应。
然而,传统物理混合催化剂存在位点分布随机、中间体传输效率低等问题。二维过渡金属硫属化合物(如MoS₂)具有面内和边缘位点空间分离的特征,且1T′相MoS₂具有独特的纯S边缘终止结构和金属性电子态,为构建单材料串联催化平台提供了理想载体。
近日,江南大学娄阳、中国科学院深圳先进技术研究院郑勇平、华东理工大学詹望成在Nature Communications发表了题为"Spatially coupled CO₂ activation and hydrogenation sites in 1T′-MoS₂ enable near-unity methanol selectivity"的研究论文。

1. 构筑了具有独特S边缘终止的氧化1T′-MoS₂催化剂,消除传统2H相的非均一活性位点。 2. 实现空间有序的面内氧取代硫缺陷与氧化边缘位点的协同作用,分别用于CO₂活化和C-O键稳定。 3. 在210°C下实现23.0%的CO₂转化率和99.2%的甲醇选择性,比反应速率达0.91 ± 0.01 gmethanol/gcat/h。 4. 揭示串联催化机理:CO₂在面内位点解离生成弱吸附*CO,经定向脱附-再捕获迁移至边缘位点完成加氢。 5. 建立单材料串联催化范式,证明原子级邻近的分级活性位点可有效调控多步加氢反应选择性。
📄 全文速览
开发高效的CO₂加氢制甲醇催化剂迫在眉睫,但精确控制中间体转移仍是挑战。该研究展示了氧化的二维1T′-MoS₂通过其固有的二维结构优势,在单一材料内独特地集成了CO₂活化和选择性加氢双重功能。
该空间有序系统在210°C下实现了23.0%的CO₂转化率、99.2%的甲醇选择性,比反应速率达0.91 ± 0.01 gmethanol/gcat/h。研究揭示了完整的反应轨迹:(i)CO₂优先在面内位点解离生成弱吸附的*CO中间体;
(ii)*CO经历定向脱附-再捕获迁移至边缘位点;(iii)1T′相的氧化边缘在加氢过程中独特地稳定C-O键。该工作确立了相工程1T′-MoS₂作为单材料串联催化的范式,展示了空间耦合活性位点如何促进CO₂加氢。
📊 图文解读
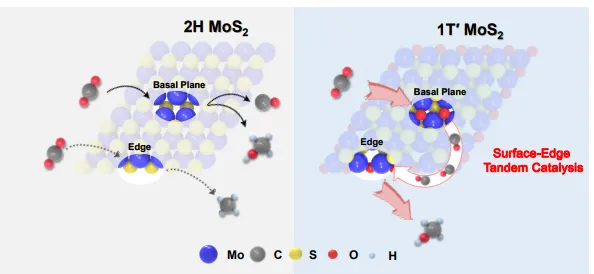
图1 | 氧化1T′-MoS₂/C(Air)催化剂的设计策略与优势示意图,展示其独特的S边缘终止、空间耦合的面内/边缘活性位点及电子结构特征,并标注关键催化性能数据。
该图阐述了氧化1T′-MoS₂催化剂的三大关键优势:独特的S边缘终止结构消除了传统2H相的非均一活性位点;空间有序的氧取代面内缺陷与氧化边缘位点协同工作;独特的电子结构可同时实现CO₂活化和C-O键稳定。
该催化体系在210°C下实现了99.2%的甲醇选择性和23.0%的CO₂转化率,比反应速率达0.91 ± 0.01 gmethanol/gcat/h。
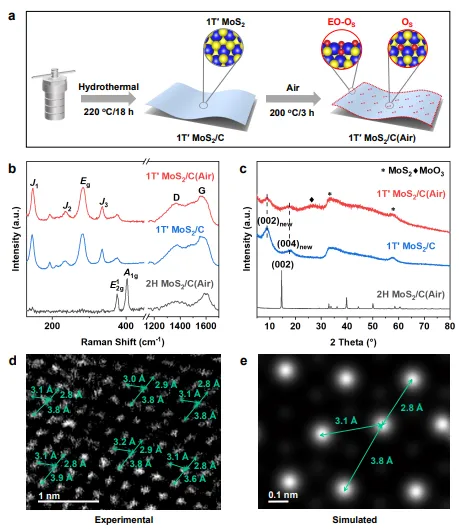
图2 | 1T′ MoS₂/C(Air)的相结构表征,包括合成路线示意图、Raman光谱、XRD图谱以及HAADF-STEM图像和模拟结果,证实其高纯度1T′相和独特的Mo原子锯齿链排列。
该图展示了通过葡萄糖辅助水热法合成1T′ MoS₂/C(Air)的路线。Raman光谱在146、234、280和334 cm⁻¹处显示1T′相特征峰(J₁、J₂、E₁g、J₃),XRD在33.5°处出现1T′特征峰,证实相纯度达96%。
HAADF-STEM图像清晰显示Mo原子呈不对称聚集,形成一维锯齿链结构,Mo-Mo间距分别为2.8 Å、3.1 Å和3.8 Å,与理论模拟一致,证实了1T′相的独特原子排列和金属性电子态。
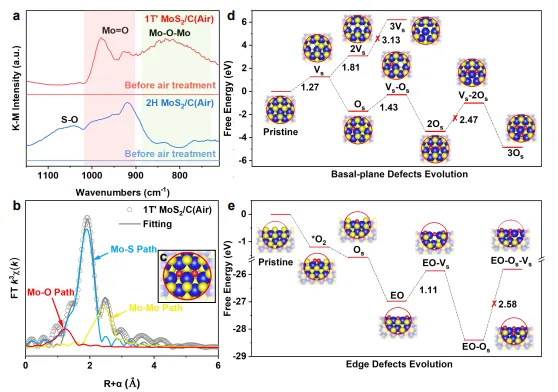
图3 | 活性位点的结构鉴定与演化机制,展示原位DRIFTS、EXAFS光谱及DFT计算结果,揭示空气氧化过程中面内氧取代硫缺陷(2OS)和边缘氧化位点(EO-OS)的形成过程。
原位DRIFTS检测到边缘Mo=O(900-1000 cm⁻¹)和表面Mo-O-Mo(700-860 cm⁻¹)物种,证实1T′相在面内和边缘均发生氧化,而2H相仅边缘氧化。
EXAFS拟合显示Mo配位环境为Mo-S₅和Mo-O₁,形成独特的氧取代硫(OS)缺陷。DFT计算表明,面内形成双氧取代硫缺陷(2OS)在热力学上有利,边缘则形成完全氧化边缘(EO)和亚边缘氧取代(EO-OS)结构。
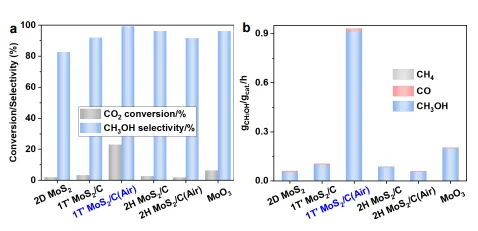
图4 | CO₂加氢催化性能评价,对比2D MoS₂、1T′ MoS₂/C、1T′ MoS₂/C(Air)、2H MoS₂/C、2H MoS₂/C(Air)和MoO₃的CO₂转化率、甲醇选择性和比反应速率。
1T′ MoS₂/C(Air)在210°C下表现出最优性能,CO₂转化率达23.0%,分别是2D MoS₂、1T′ MoS₂/C、2H MoS₂/C、2H MoS₂/C(Air)和MoO₃的11.6、6.9、8.5、12.4和3.6倍。
比反应速率达0.91 ± 0.01 gmethanol/gcat/h,是其他催化剂的4.6-17.1倍,且远高于文献报道的>99%选择性催化剂(通常≤0.1 gmethanol/gcat/h)。
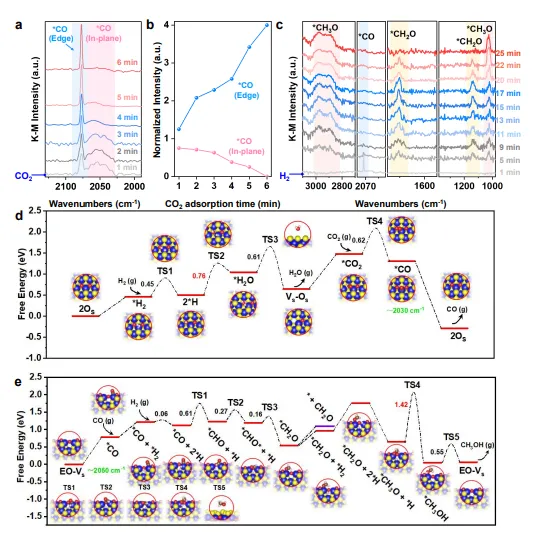
图5 | 基于DFT计算的反应机理研究,展示CO₂在面内2OS位点解离、*CO中间体迁移至边缘EO-OS位点、以及在边缘位点逐步加氢生成甲醇的完整反应路径和能量变化。
该图通过DFT计算揭示了空间耦合活性位点的串联催化机理。CO₂优先在面内双氧取代硫(2OS)缺陷位点解离,生成弱吸附的*CO中间体,该过程具有较低的能量壁垒。
*CO随后经历定向脱附-再捕获过程迁移至边缘氧化位点(EO-OS),避免了C-O键过早断裂。在边缘位点,氧化的1T′相结构独特地稳定了C-O键,通过多步加氢最终生成甲醇,而非甲烷副产物。
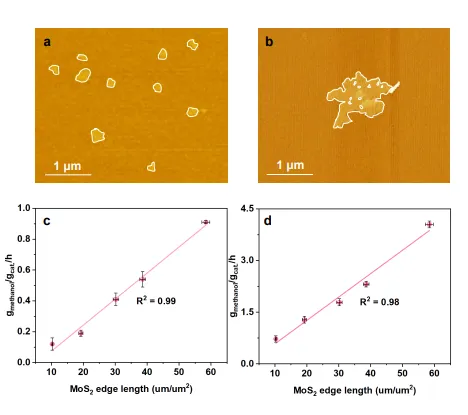
图6 | 催化剂的稳定性评估及串联催化概念验证,展示1T′ MoS₂/C(Air)在长时间运行中的性能保持率,以及空间耦合位点与物理混合催化剂的性能对比。
与机械混合的1T′ MoS₂/C+MoO₃催化剂相比,1T′ MoS₂/C(Air)的CO₂转化率提高了约8.2倍,甲醇选择性从90.5%提升至99.2%,证明了原子级空间邻近的活性位点相比物理混合具有显著优势。此外,该催化剂在长时间运行中保持良好的性能稳定性,证实了其作为单材料串联催化平台的实用潜力。
📝 总结
该研究通过相工程策略开发了氧化的1T′-MoS₂催化剂,成功实现了CO₂加氢制甲醇的高活性和近100%选择性。该催化剂的面内氧取代硫缺陷和氧化边缘位点构成了空间耦合的串联活性中心,分别负责CO₂活化和C-O键稳定。
机理研究揭示了从CO₂解离到*CO迁移再到加氢的完整反应路径。这项工作为单材料串联催化提供了新范式,展示了通过精确调控二维材料表面位点空间分布来优化多步催化反应的可行性。
Spatially coupled CO₂ activation and hydrogenation sites in 1T′-MoS₂ enable near-unity methanol selectivity,Nature Communications,2026,DOI:10.1038/s41467-026-72205-1
#江南大学#娄阳#中国科学院深圳先进技术研究院#郑勇平#华东理工大学#詹望成#催化#Nature子刊

随机文章
-
 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
- 深圳湾实验室传染病研究所——病毒传染病课题组招聘公告
- 直属第四、第五党支部联合开展参观深圳市垃圾处理历史博物馆学习活动
- 深圳将打造防治肿瘤个性化医学前沿阵地:个性化医学“医-教-研-用”(南方)科技中心在成立
- 茶颜悦色深圳开业,黄牛炒到88元一杯!
- 深圳稳工好厂推荐:【纯27元/H】不过安检/可带手机/个人岗位!
- 每周五及五一假期深圳起止厦门/东山岛跟团3天旅游团698,厦门鼓浪屿、南普陀寺、厦大、曾厝垵、东山岛南门湾、漳州古城、苏峰山环岛路
- 官宣!陈慧娴40周年深圳站正式定档
- 深圳5区体育中考人数激增6497人!2026年中考生总人数预测出炉
- 深圳海科船舶固定YT-98H-M4四合一应用案例
- 五一假期深圳起止桂林阳朔跟团3天598,聚龙潭,多人竹筏,少数民族古寨,十里画廊骑行,兴坪古镇20元人民币背景,阳朔西街,印象刘三姐
