汪国秀院士领衔!深圳理工大学郭鑫最新AM | 自调节钠离子电池材料:从相重构到功能激活!
- 2026-06-26 16:29:21
汪国秀院士领衔!深圳理工大学郭鑫最新AM | 自调节钠离子电池材料:从相重构到功能激活!
【做计算 找华算】新学期科研提速!华算科技预存增值高至30%,送¥8500+返利!额满即止,快来抢占! 钠离子电池正朝着实际部署迈进,然而其长期耐用性并非由静态的材料属性决定,而是由晶格、界面和电解质环境在运行条件下的耦合演化所主导。然而,目前仍缺乏对这些分散的机理见解的系统性整合。 2026年3月10日,悉尼科技大学汪国秀、Hao Liu、深圳理工大学郭鑫在国际知名期刊Advanced Materials发表题为《Self-Regulating Sodium-Ion Battery Materials: From Phase Reconstruction to Functional Activation》的综述论文,Hong Gao、Dingyi Zhang、Chao Wang为论文共同第一作者,汪国秀、Hao Liu、郭鑫为论文共同通讯作者。 
本综述引入了一个统一的自调节框架,在该框架中,结构和化学变化被限制在可逆且功能上与电化学传输保持一致的范围内。证据围绕三个主线进行组织:层状氧化物中的晶格与相演化,其中受控的层间滑移、缓和的钠-空位有序化以及稳定的氧参与收窄了相变窗口;界面化学与力学,强调富含无机物、可自我更新的界面结构,这些结构能够维持离子传输并限制溶解;以及电解质-材料耦合,其中溶剂化结构、浓度区间和靶向添加剂引导界面重构和近表面传输。范围涵盖了主要的正极材料体系和自缓冲负极,并将电解质视为有目的的促成因素。基于这些见解,作者提炼出将原位特征与成分和工艺选择联系起来的设计准则,并概述了模型指导的优化和数据驱动的发现所带来的机遇。该框架为实现可在实际约束下可靠运行的可编程、耐用钠离子电池提供了一个以材料为先的路线图。 由于钠资源丰富,前驱体供应链灵活,且现有的锂离子制造工艺只需有限的改造即可适用,钠离子电池(SIBs)在电网和成本敏感型储能领域正获得越来越多的关注。决定电池耐用性的关键因素仍然是活性材料及其界面在运行过程中的演化,而非其静态属性。在本综述中,“自调节”指在循环过程中保持受限和自限制的结构与化学演化,从而使传输路径保持开放,副反应受到抑制,性能在长期使用中得以维持。本综述聚焦于采用层状氧化物正极和自缓冲合金或转化型负极的钠离子电池。电解质被视为用于强化预期材料行为的靶向促成因素。在此范围内,作者沿着四个耦合的主线来组织该领域。首先,层状氧化物中的晶格-相轴:一个O3↔P3相变窗口,其中O2相仅在深度脱钠时出现,加上受抑制的钠-空位有序化和受控的层间滑移,实现了低滞后循环,而成分、位点和熵设计则引导着这些路径。其次,界面轴:基于无机骨架并由薄有机层覆盖的稳健的正极电解质界面(CEI)和固体电解质界面(SEI),能够保持电荷转移和扩散通道,并抑制气体产生和过渡金属溶解。第三,由成分-结构编程的本征自修复:通过联合调节电极成分(包括位点/熵设计)和自缓冲结构,电极可以将相演化和界面/机械降解限制在一个可逆的窗口内,从而在不依赖外部触发的情况下,在循环过程中维持贯通的离子/电子传输。第四,电解质-材料耦合:溶剂化状态和浓度区间引导界面化学和近表面传输,放大了由晶格和界面编码的可逆性。 最近研究的证据支持了自调节在钠离子电池中的重要性。例如,在层状氧化物正极中,熵工程和位点设计可以稳定晶格氧的参与,同时最小化滞后并平滑相演化。在负极方面,蛋黄-壳结构的Sb@Void@石墨炔纳米盒体现了本征自调节:内部空隙缓冲了Sb的膨胀,π共轭的石墨炔壳层维持了电子/离子的贯通,并且SEI在反复重构过程中保持致密。在全电池(Sb@Void@石墨炔 ∥Na3V2(PO4)3)中,在1 A g⁻¹下循环500次后,容量仍保持在354 mAh g⁻¹。此外,电解质添加剂可以在界面处触发自调节。例如,二氟(草酸)硼酸钠(NaDFOB)将SEI重构为一个薄的有机表层,其下是一个富含NaF和硼酸盐物种的致密子层,该子层在前约50次循环中变得更加致密。研究表明,这种演化能够密封界面缺陷,限制溶剂接触,并维持离子传导。这种行为代表了一种自钝化的原型,并支持了钠离子电池的耐用循环。综上所述,自调节作为一种设计理念,将晶格路径与界面更新对齐,并选择能够加强这种对齐的电解质。 尽管取得了进展,但仍有几个实际问题尚待解决。在高面容量的电极中,界面必须在曲折的介观结构中快速成核并保持空间均匀,以确保电荷转移和扩散路径的畅通。应在保持高电压运行和可接受的初始效率的同时,抑制溶解的过渡金属物种与负极界面之间的串扰。要在宽温度范围内可靠使用,需要界面能够在低温下防止溶剂共嵌入,并在高温下抑制加速的副反应。最后,设计准则需要在规模化生产的约束下(包括粘结剂化学、导电网络结构、辊压和堆叠压力)保持有效,以便在纽扣电池中展示的行为能够转化为工业化电极。 基于这一理论基础,本综述的其余部分沿着示意图1中总结的四个耦合主线汇集了原位证据:(i)可编程的晶格/相轨迹,(ii)自限制的界面化学与力学,(iii)由成分/结构引导的本征自修复(包括自缓冲负极结构),以及(iv)强化预期的晶格-界面反馈的电解质-材料耦合。在此路线图中,作者将机理特征转化为层状正极的成分、位点和熵策略,并考察自缓冲负极,重点关注颗粒尺寸、导电网络结构和应变耗散。 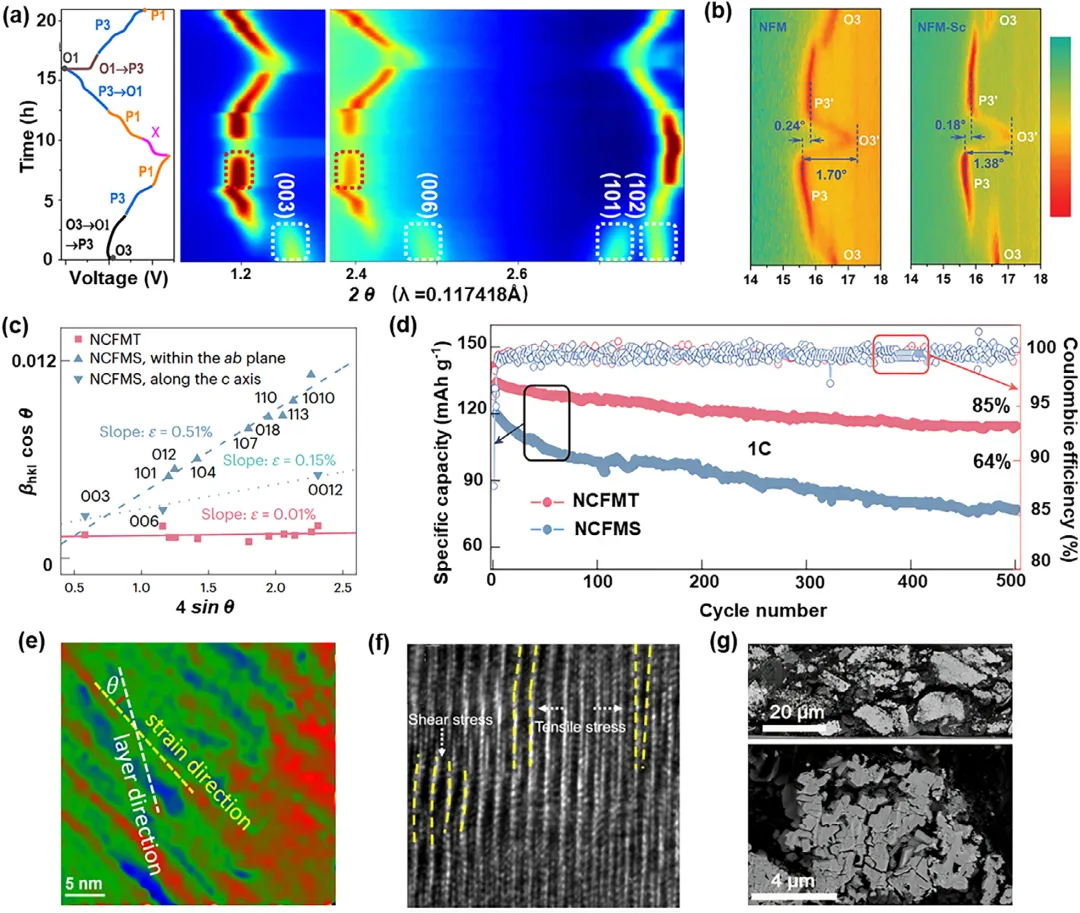
图1:a) O3型NaNi0.4Mn0.4Co0.2O2正极在2.0至4.4V电压范围内充放电过程中的电压曲线及相应的原位同步辐射X射线衍射(SXRD)图谱的二维等高线图;颜色表示衍射强度(红色最高,蓝色最低)。b) 在2.0至4.2V电压范围内的初始充放电过程中,来自原位XRD的(003)衍射峰的等高线图。c) Williamson–Hall分析得出的微观应变(ε);NCFMS中的各向异性是通过对主要由面内(ab;虚线)或面外(c;点线)贡献的hkl衍射峰组进行独立拟合获得的。d) 室温循环性能。e) 原始O3-NaNi0.4Mn0.4Co0.2O2的几何相分析(GPA)。f) 高倍透射电镜图像中虚线方框区域的放大图。g) 循环200次后NFMMT的扫描电镜(SEM)图像。 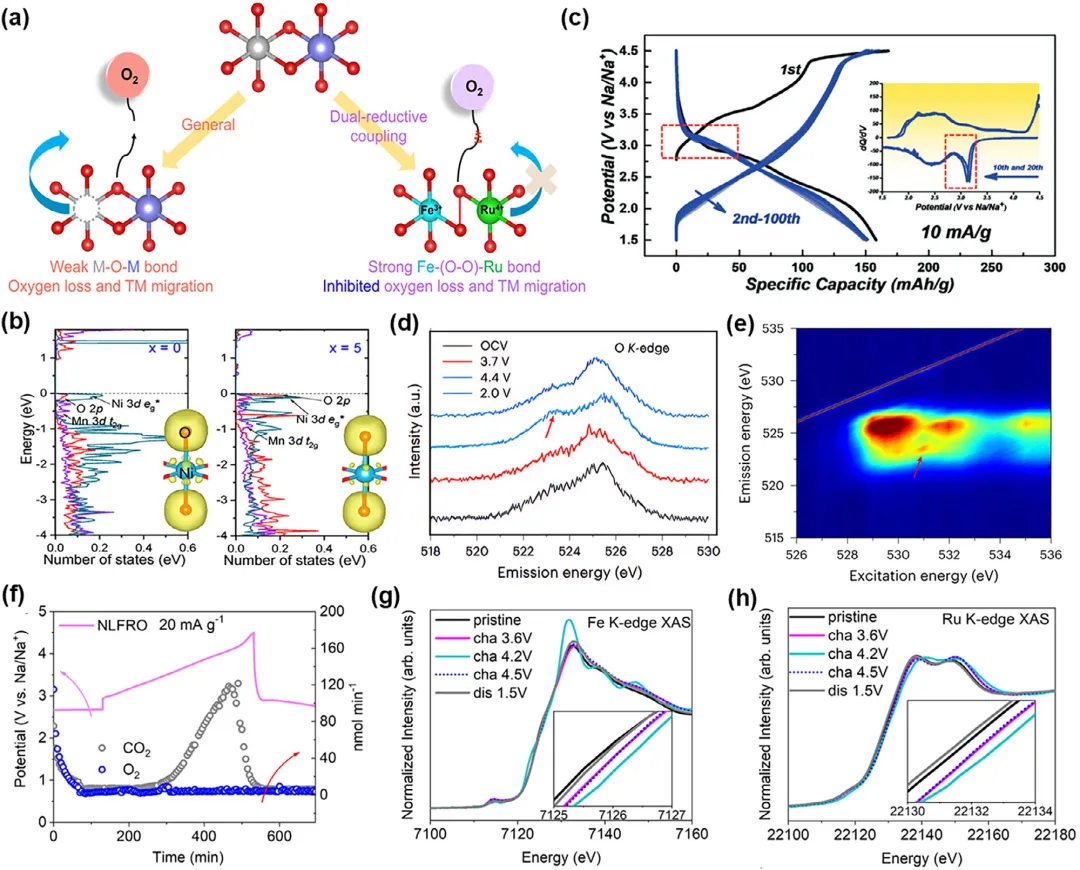
图2:a) 示意图,展示了强过渡金属-氧(TM─O)共价键稳定氧化态氧物种。b) NLRO在1.5至4.5V电压范围内,10mA g⁻¹电流密度下的典型充放电曲线;插图:第10圈和第20圈的dQ/dV图。c) 部分态密度(pDOS)图,显示了x=0时Ni 3d eg*、Mn 3d t2g和O 2p轨道的演化。d) 在531.0 eV激发下的非原位氧K边共振非弹性X射线散射(RIXS)谱图;箭头表示晶格氧氧化还原特征。e) 充电至4.4V样品的氧K边mRIXS谱图。红色箭头指示的额外峰表明氧参与了完全充电状态下的氧化还原。f) 循环过程中的原位差分电化学质谱(DEMS)。g) Fe和h) Ru K边X射线吸收近边结构(XANES)谱在初始循环过程中的变化。 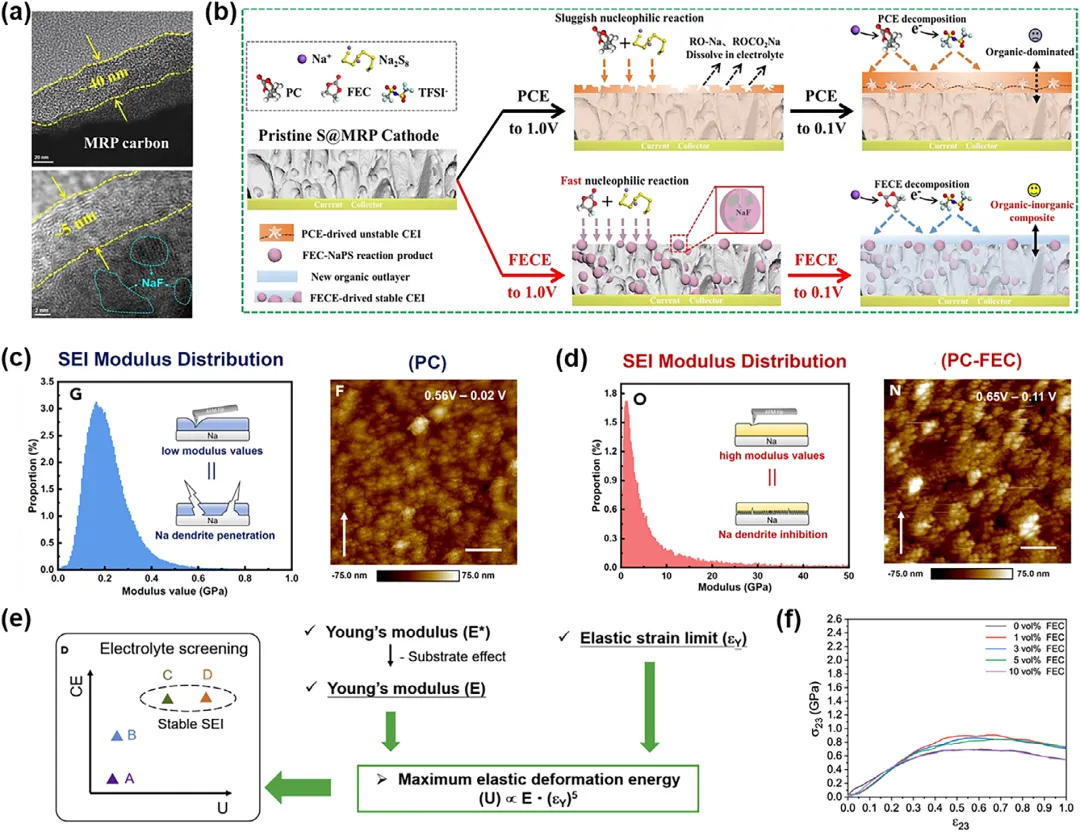
图3:在不同电解质的S@MRP||Na电池放电至0.1V时,正极电解质界面(CEI)的微观结构和成分。a) PCE(上)和FECE(下)电解质中CEI的透射电子显微镜(TEM)分析。b) S@MRP||Na电池在两种电解质中形成完整CEI的机理示意图。c) PC电解质中形成的SEI模量值概率分布(左);原子力显微镜(AFM)检测PC电解质中铜集流体上SEI的最终形貌(右)。d) PC-FEC电解质中形成的SEI模量值概率分布(左);AFM检测PC-FEC电解质中铜集流体上SEI的最终形貌(右)(比例尺:1 µm)。e) 最大弹性形变能U与库仑效率的对比总结柱状图。f) 在0.0、1.0、3.0、5.0和10.0 vol % FEC条件下,从SEI膜的应力-应变曲线导出的剪切弹性常数C44(在Voigt表示法中为C55,zx平面纯剪切),显示在≈1 vol %处出现峰值,对应于具有最大抗剪切能力的致密膜。 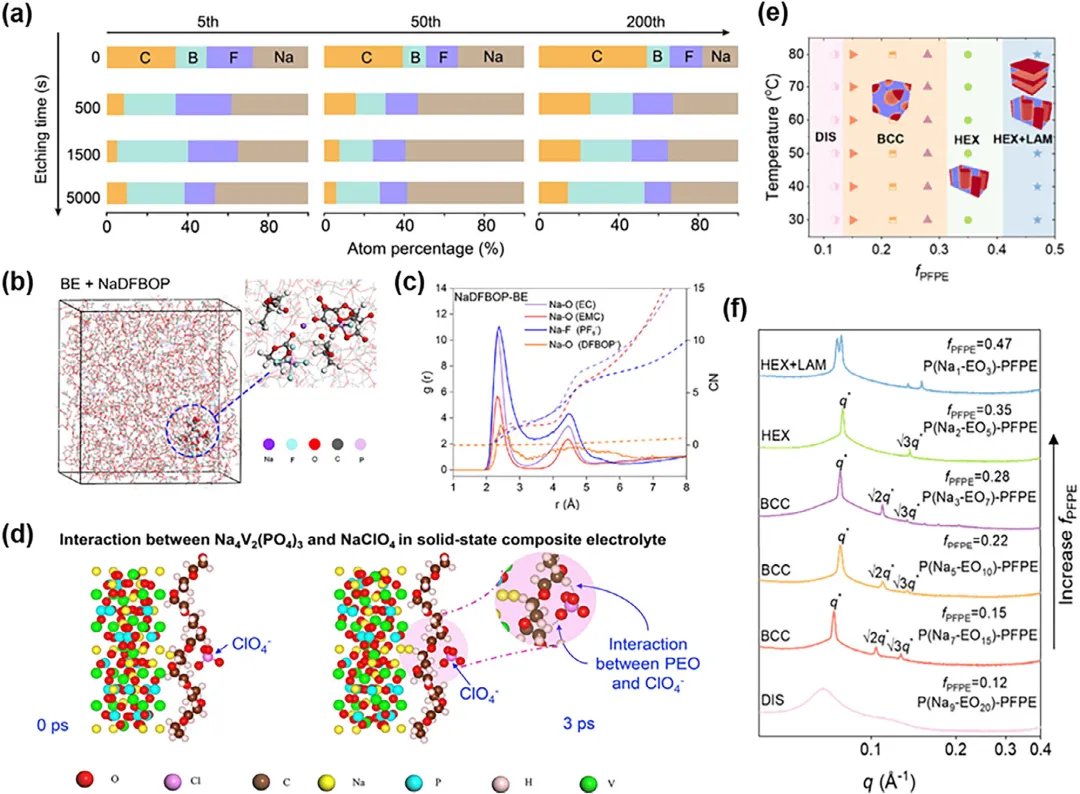
图4:a) NaDFOB衍生的固体电解质界面(SEI)的XPS深度剖面元素丰度(第5、50、200次循环)。第50次循环的膜在内层表现出最高的Na和无机(B、F)含量,证明了其致密、缺陷密封的钝化框架。b) MD模拟快照和c) BE + NaDFBOP中Na+的径向分布函数(RDF)曲线和配位数曲线。d) 模拟的全固态铁电工程复合电解质中NVP负极与NaClO₄之间的相互作用。e) P(Nam-EOn)-PFPE的相图,总结了在不同温度和fPFPE下不同的自组装形貌。插图提供了每种相应纳米结构的示意图。红色区域代表PFPE域,而蓝色区域代表PEO和离子域。f) 在80℃下,具有不同fPFPE的P(Nam-EOn)-PFPE的同步辐射SAXS谱图。与结构对应的布拉格峰用散射矢量q标记(q*是主散射峰的位置)。 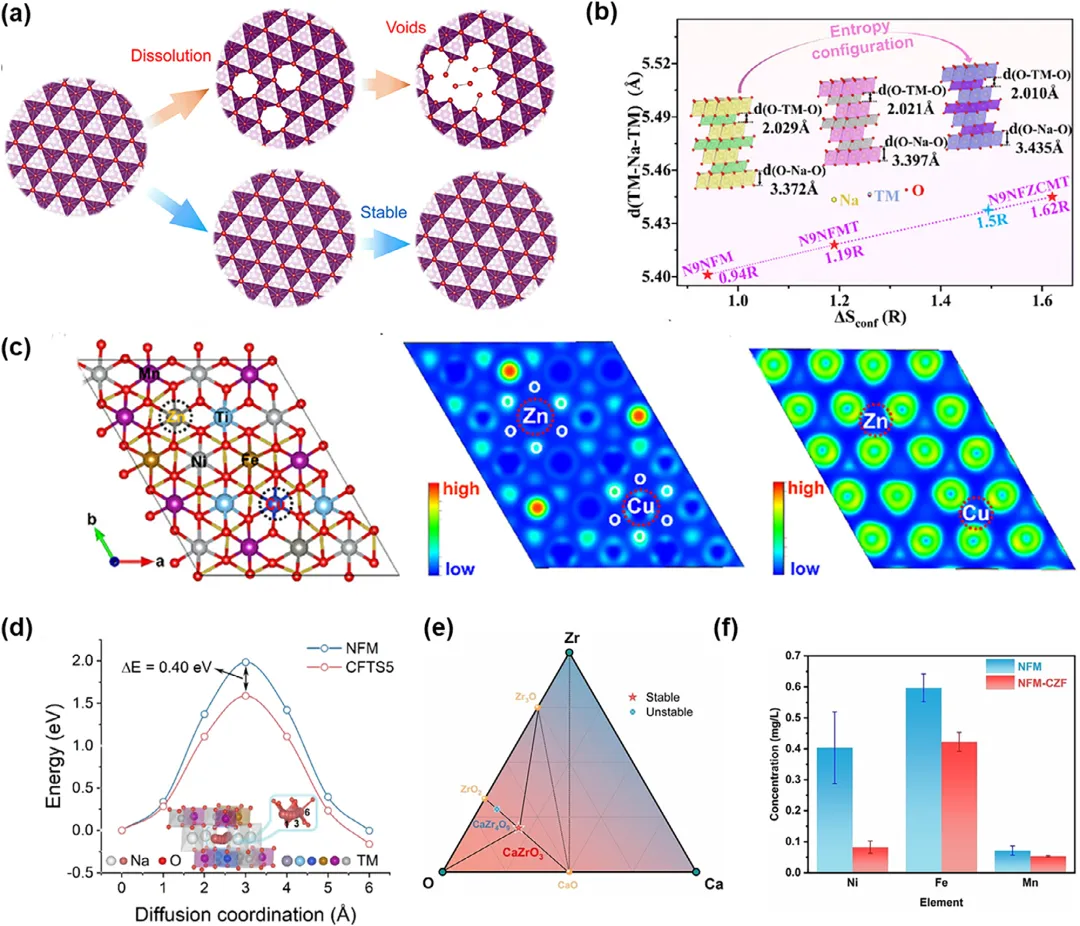
图5:a) Na0.67Mn0.57Ni0.28Fe0.15O2正极中空洞形成和具有增强结构稳定性的高熵氧化物(HEO)的示意图。b) 构型熵(∆Sconf)与O-Na-O板层间距的变化趋势。c) N9NFZCMT的优化晶体结构,及其沿着TM层(001)和氧层(001)晶格平面的价电子的电子局域函数(ELF)。d) NFM和CFTS5的Na⁺扩散能垒。e) Ca-Zr-O三元相图,显示了CaZrO₃的形成。f) 循环后样品的过渡金属离子溶解结果。 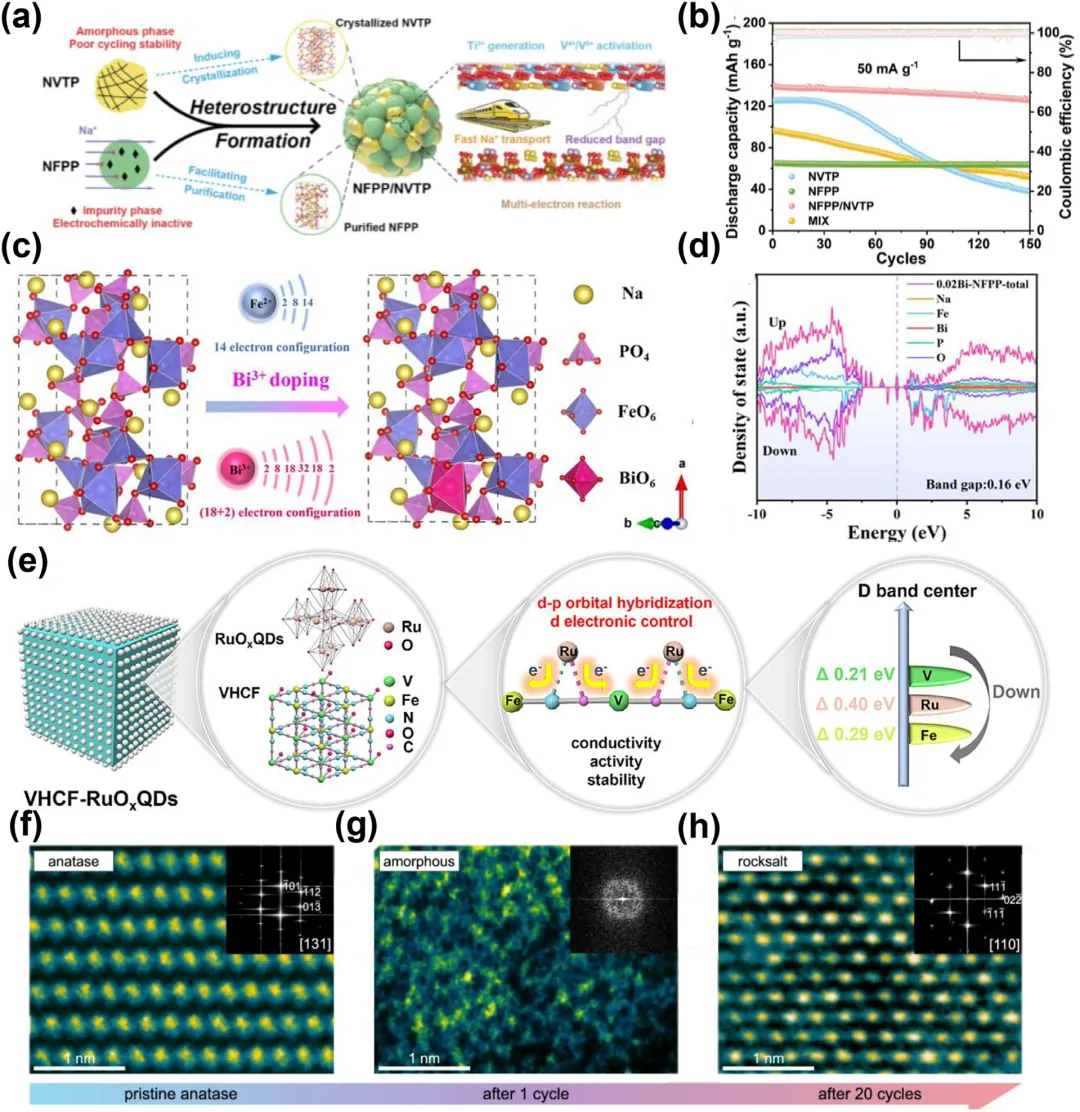
图6:a) 示意图,说明了NFPP/NVTP中的协同效应。b) 所制备正极在50 mA g⁻¹下的循环性能。c) Bi³⁺掺杂前后的晶体结构图,以及Bi³⁺和Fe²⁺离子结构的示意图。d) 0.02Bi-NFPP的态密度(DOS)。e) “原位共沉淀合成”方法制备VHCF-RuOxQDs的示意图,以及VHCF和VHCF-RuOxQDs的三级放大的局部结构图。f) 原始锐钛矿相、g) 循环1次后和h) 循环20次后的高角度环形暗场扫描透射电子显微镜(HAADF-STEM)图像及内嵌的快速傅里叶变换(FFT)图谱,显示了局部原子排列的演变和c-a'-RS相变过程。 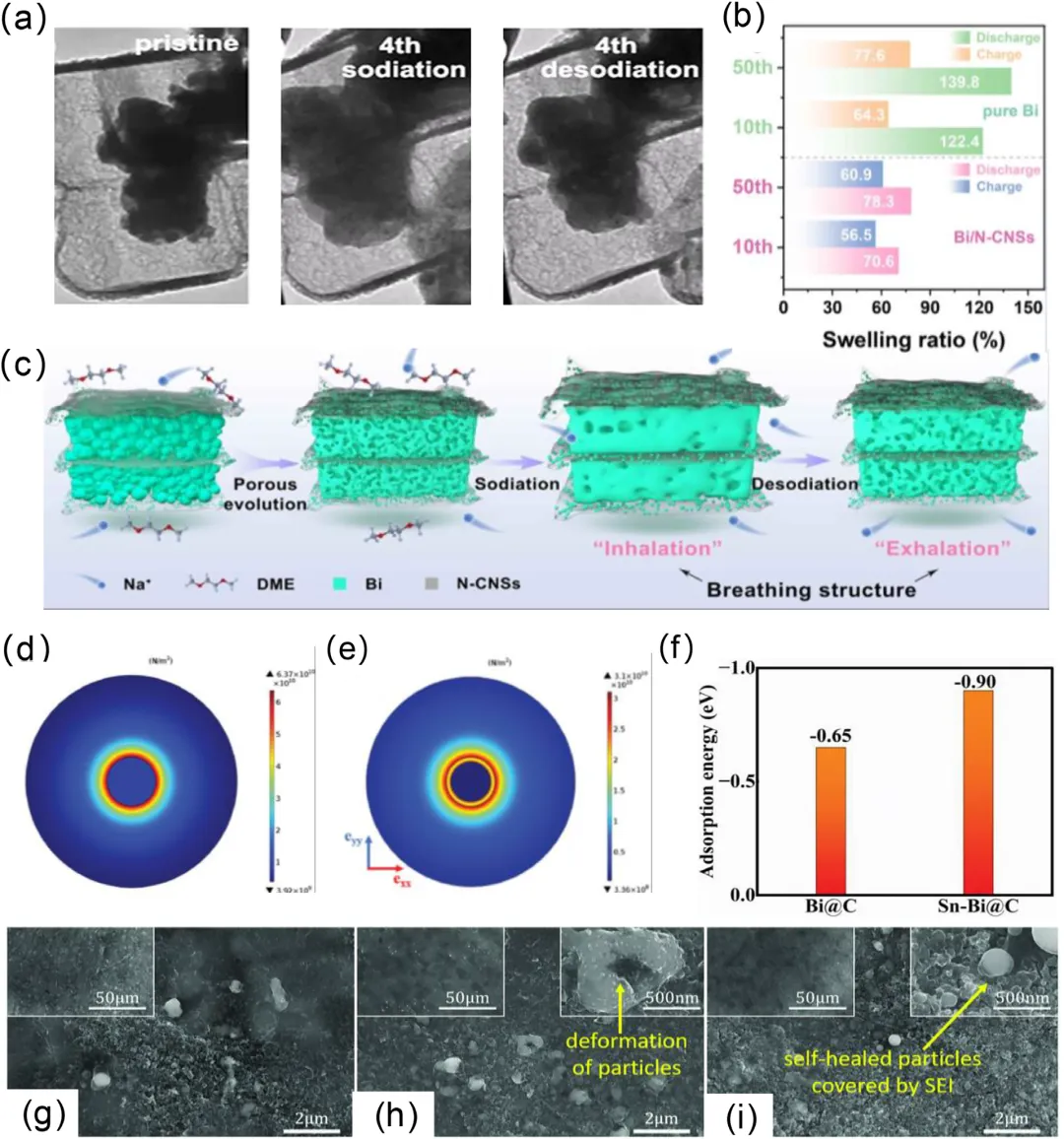
图7:a) 在原位透射电子显微镜(TEM)中估算Sb核的体积膨胀率。b) 完全充电/放电的Bi/N-CNSs和纯Bi电极在不同循环次数后的膨胀率。c) Bi/N-CNSs在循环过程中形态演变和“呼吸”行为的示意图。d, e) 通过有限元模拟分析的d) Sn@C和e) Sn-Bi@C在完全钠化状态下的应力大小和分布。基于DFT的理论计算。f) Sn-Bi@C和Bi@C中Na吸附能的比较。g-i) 半电池中液态金属纳米颗粒(LMNPs)电极在g) 完全合金化状态、h) 萃取过程中和i) 完全萃取状态下的扫描电镜(SEM)图像。每张图像的插图与主图像属于同一一样品。电化学测试以10C的倍率进行。 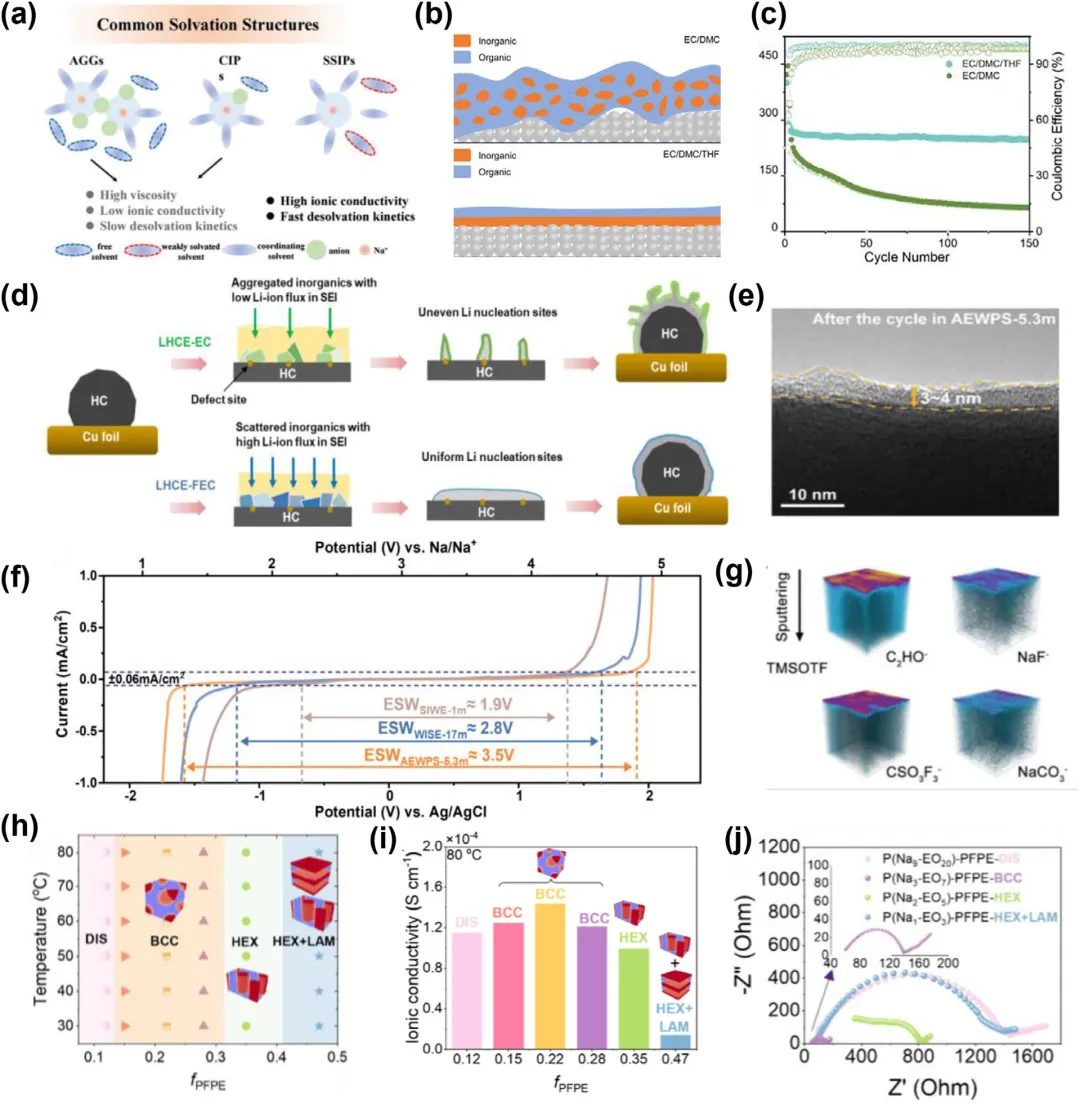
图8:a) Na⁺周围的主要溶剂化结构:离子聚集体(AGGs)、接触离子对(CIPs)和溶剂分离离子对(SSIPs)。b) 在不同电解质中形成的固体电解质界面(SEI)结构示意图。c) EC/DMC和EC/DMC/THF电解质在1C倍率下的长期循环性能。d) 示意图,展示了在局部高浓度电解质(LHCEs)中,EC和FEC对硬碳(HC)上SEI化学和镀层形貌的影响。e) 在AEWPS-5.3 m电解质中循环后PW的高分辨率透射电镜图像。f) 不同电解质在10 mV s⁻¹扫描速率下的线性扫描伏安法曲线。g) 来自TMSOTF的循环后HC电极的飞行时间二次离子质谱(TOF-SIMS)图像。h) P(Nam-EOn)-PFPE的相图,总结了在不同温度和fPFPE下的不同自组装形貌。插图提供了每种相应纳米结构的示意图。i) 在80℃下,P(Nam-EOn)-PFPE电解质的离子电导率随fPFPE组成的变化。j) 半圆曲线表示在80℃下循环160小时后电池的电阻。 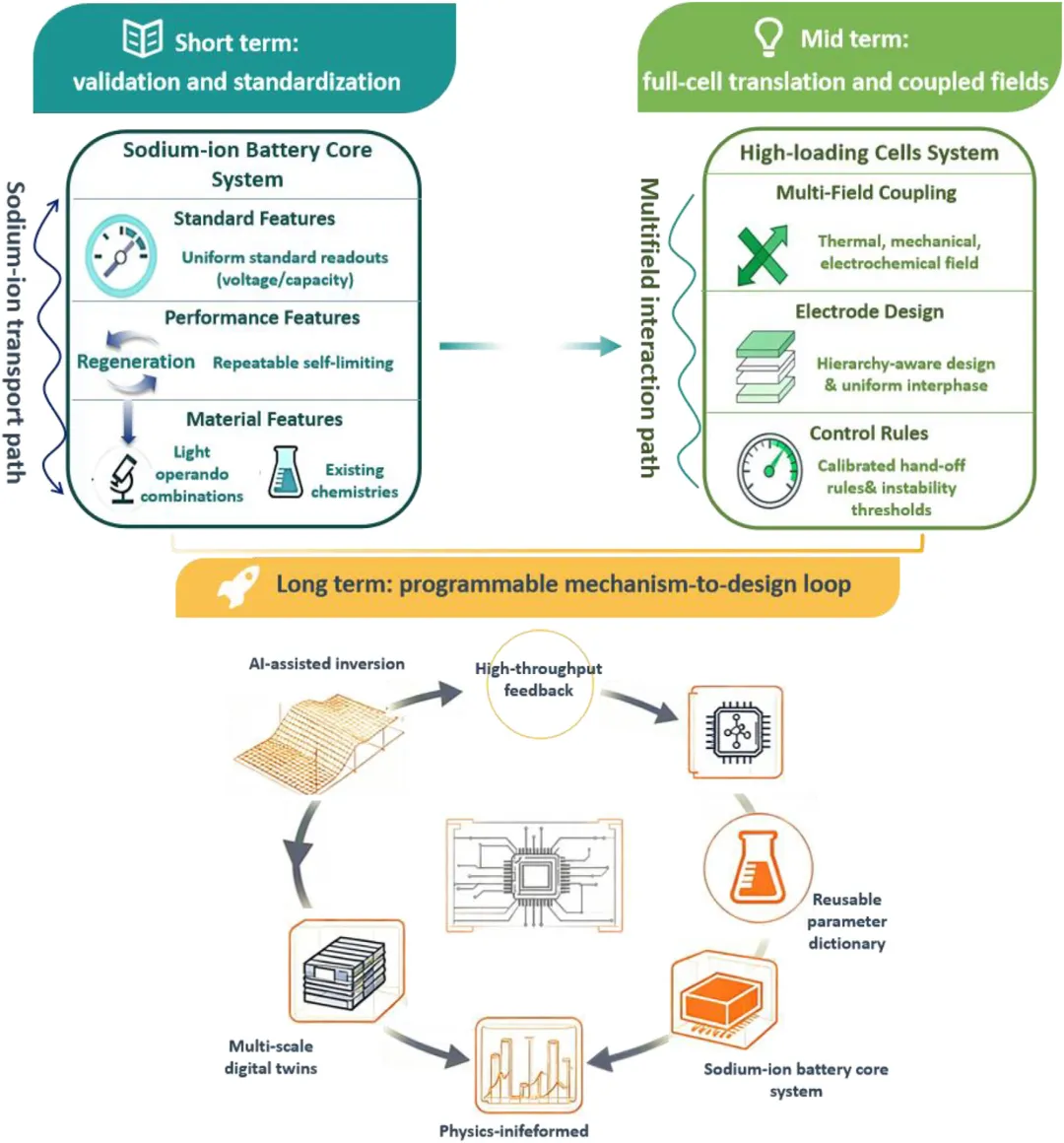
图9:在钠离子电池中实现原位自调节的时间尺度路线图和闭环工作流程。 基于上文建立的框架,本节总结了本综述的核心信息,并概述了在实践中实施自调节的时间尺度路线图。自调节的证据基础与引言中强调的四个耦合路径保持一致:(i)晶格/相演化,(ii)界面化学与力学,(iii)用于本征自修复的成分和结构,以及(iv)电解质-材料耦合。在这些路径中,实际目标不是扩大化学范围,而是建立可比较的验证、可转移的读出指标和明确的交接规则,以保持从半电池到高负载全电池的机理意义。 几个未解决的问题继续限制着清晰度和转化应用。由于可逆与不可逆演化机制的定义和报告不一致,机理的可比性仍然受到限制,尤其是在相轨迹、界面更新和传输衰减共存的情况下。界面化学-力学性质的描述仍然常常是定性的,界面连续性、应力适应以及贯通传输路径的持久性之间的联系通常没有以可转移的方式进行参数化。电解质效应的归因并不总是足够严谨,支持性耦合与外部施加的钝化各自的作用也未能通过证据链得到一致的区分。从半电池到全电池的转化仍然是一个主要瓶颈,因为在稀释条件下观察到的特征在贫电解液、高面载量、堆叠压力和热梯度下通常会发生变化。 这些差距促使作者制定了下述的短期、中期和长期路线图,该路线图保持了固定的化学范围,并有选择地使用电解质来加强晶格演化和界面更新之间的耦合。 近期的优先事项是统一一套标准的读出指标和透明的报告方式。形成和稳态库仑效率、电荷转移和质量传输的阻抗与弛豫特征、间歇滴定的扩散趋势,以及充放电开路电压曲线之间的面积,构成了一个充分的基础;在有信息价值的情况下,可以加入原位气体分析和过渡金属串扰分析。使用现有化学体系,研究应通过轻量级的原位组合分析和明确规定的工艺参数(如化成方案、温度和上限电压窗口)来展示可重复的自限制行为和再生能力。成功的评判标准是跨实验室的可重复性,而不是单一指标的极端值。 中期目标是在耦合的热、机械和电化学条件下,评估高负载全电池的转化应用。在化学体系固定的情况下,具有层级意识的电极和电池设计——孔隙率控制、导电网络连续性、堆叠压力和热管理——应能在保持传输路径开放的同时,实现快速且空间均匀的界面形成。根据标准读出指标校准的紧凑模型可以通过定位不稳定性阈值并指示哪些操作参数可恢复可逆循环,从而为从半电池证据到全电池操作提供实用的交接规则。 在更长远的未来,人工智能辅助的反演、预测和优化可以使“机制到设计”的循环变得可编程。标准读出指标可以映射到可解释的演化参数,如随时间变化的相窗口宽度、层间滑移动力学、界面连续性和传输系数。带有不确定性量化的物理信息代理模型可以预测对工艺参数的响应,并识别在实际约束下保持可逆行为的操作模式。与此同时,多尺度数字孪生和高通量实验通过更新模型和整合可复用的参数字典来闭合循环,从而在不改变底层化学体系的情况下实现规模化生产,并向着材料系统的可编程、自演化控制迈进。 Self-Regulating Sodium-Ion Battery Materials: From Phase Reconstruction to Functional Activation. Adv. Mater., 2026. https://doi.org/10.1002/adma.202523596. #悉尼科技大学#汪国秀#深圳理工大学#郭鑫#电池#AM 👉 点击阅读原文,立即下单!

本文来自网友投稿或网络内容,如有侵犯您的权益请联系我们删除,联系邮箱:wyl860211@qq.com 。

