
辞旧迎新,知识续航!「龙哥读论文」陪你跨年,知识星球会员优惠券限时限量放送!🐉 「龙哥读论文」知识星球:让你看论文像刷视频一样简单!公众号每日8篇拆解不够看?星球无上限更AI领域论文、资讯、招聘、招博、开源代码,一站式干货,每日2分钟刷完即赚!👇扫码加入「龙哥读论文」知识星球,前沿干货、实用资源一站式拿捏~

龙哥推荐理由:
这篇论文解决了一个非常实际且重要的问题:如何用低成本的低分辨率空间转录组数据,“脑补”出高分辨率的基因表达图谱。它不仅提出了一个性能强悍的模型,更重要的是,它从根本上重新思考了任务的定义,引入了物理守恒定律作为硬约束,并实现了令人印象深刻的跨物种、跨平台的零样本泛化。对于AI在生物医学领域的落地应用,这种兼具理论严谨性和实用性的工作,非常值得一读。
原论文信息如下:
论文标题:
Towards Universal Spatial Transcriptomics Super-Resolution: A Generalist Physically Consistent Flow Matching Framework
发表日期:
2026年02月
发表单位:
深圳市人民医院,哈尔滨工业大学(深圳),大湾区大学,深圳大学
原文链接:
https://arxiv.org/pdf/2602.10644v1.pdf
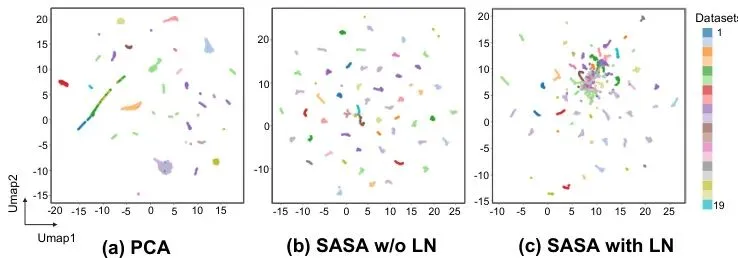
想象一下,给你一张马赛克画质的世界地图,让你“脑补”出每个国家的详细地形和资源分布——这差不多就是空间转录组超分辨率在做的事。科学家们想看清组织里每个“小格子”的基因活动,但技术又贵又糙,得到的往往是模糊的“低分辨率”数据。以前的AI模型试过“脑补”,但一换组织类型(比如从人肝换到鼠脑)就傻眼,预测结果还经常违反“物质守恒”这种基本物理定律。🤨今天要聊的这篇论文,来自深圳市人民医院、哈工大(深圳)等团队,他们整了个叫SRast的“通用求解器”。它不仅性能强悍,更关键的是,它用一套“解耦”+“物理约束”的组合拳,从根本上解决了泛化差和结果“不物理”的两大痛点。更牛的是,它学会了“零样本”跨物种、跨平台推理——训练时见过人和老鼠的数据,没见过的斑马鱼组织切片拿过来,照样能给你“脑补”得明明白白。这泛化能力,有点东西。图2:不同数据集的特征可视化对比。(a) PCA特征,(b) SASA阶段编码后的特征,(c) 经过Latent Norm归一化后的特征。可以看到(c)中不同来源的数据点混合得更好,说明跨样本的分布差异被有效对齐了。突破瓶颈:空间转录组超分辨率的通用求解器
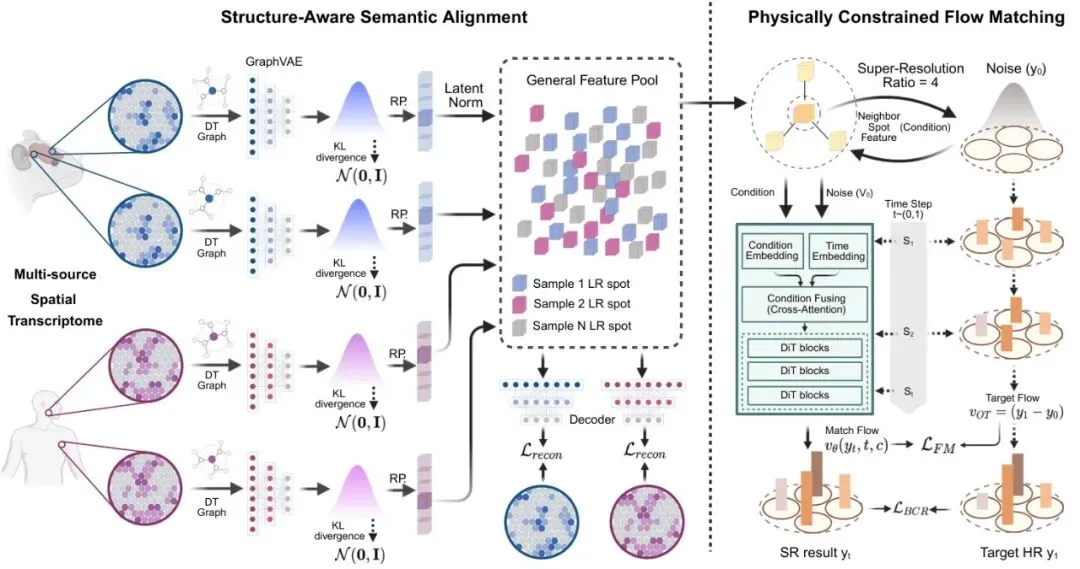
先快速科普下空间转录组学。传统测序技术好比把一杯混合果汁拿去分析成分,能知道有哪些水果,但不知道苹果块和橙子瓣在杯子里的具体位置。空间转录组技术厉害了,它相当于给这杯果汁拍了一张“基因表达分布图”,告诉你哪个位置苹果基因活跃,哪个位置橙子基因在喊叫。这对于理解器官发育、肿瘤微环境等至关重要。但高分辨率图谱成本极高。于是,空间转录组超分辨率技术应运而生:用算法从低分辨率(一个大格子)数据,推测出高分辨率(拆成多个小格子)的基因表达。1. 泛化能力差:生物数据异质性极强。不同物种、不同个体、甚至同一器官的不同切片,其基因表达模式都可能天差地别。一个在人类肝脏数据上训练得炉火纯青的模型,拿到老鼠大脑数据上,性能可能断崖式下跌。这属于典型的分布外泛化问题。2. 缺乏物理一致性:这是更根本的问题。一个低分辨率格子测到的总RNA量,必须等于它拆分出的所有高分辨率小格子的RNA量之和(局部质量守恒)。但以前的方法把超分辨率当成普通回归任务,预测出的数值加总后经常对不上原始值,产生了“无中生有”或“凭空消失”的RNA,这在生物学上是说不通的。SRast就是为了成为解决这两个核心难题的“通用求解器”而被设计出来的。它的核心思路可以用下面这张总览图一目了然:图1:SRast框架总览。(左)结构感知语义对齐(SASA):使用样本特定的图变分自编码器,基于双拓扑图生成样本表示,并利用潜在归一化消除多源数据的批次效应,构建统一的通用特征池。(右)物理约束流匹配(PCFM):基于DiT架构,注入通用特征及其邻域特征作为条件,通过流匹配学习从噪声到高分辨率比率的优化传输流,并利用KL散度约束确保生成分布与目标高分辨率分布对齐。解耦的艺术:分离语义与几何以对抗异质性
SRast的第一个大招叫结构感知语义对齐。这个名字有点拗口,但思想很直接:把“基因是什么”和“基因怎么分布”这两件事分开学。以前的方法把这两个任务耦合在一起,导致模型容易过拟合到特定组织样本的“样子”上。比如,它在人类肝脏数据里学到“A基因喜欢和B基因扎堆出现”,但这可能只是肝脏特有的模式(伪相关),换到大脑里就不成立了。首先,它利用一个双拓扑图来建模每个低分辨率数据点(spot)。这个图同时捕捉两种关系:语义邻居:基因表达模式相似的点(即使物理距离远)。(公式:双拓扑图的边集是空间近邻和语义近邻的并集)然后,用一个图变分自编码器对这个图进行编码,得到每个点的特征表示。最关键的一步来了:潜在归一化。它对所有样本的特征进行标准化,抹平不同样本、不同批次之间的分布差异(比如均值、方差的差异)。这就像是把来自不同工厂、不同标号的螺丝,都加工成统一的标准件。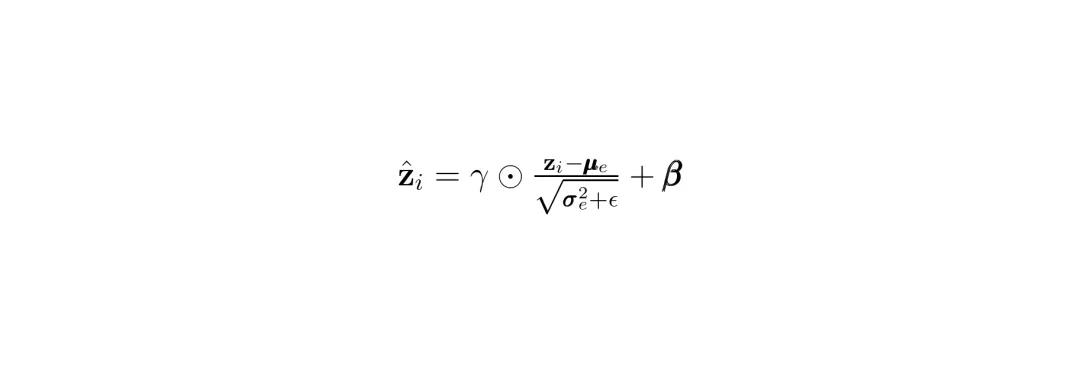 经过这一系列操作,SASA模块产出了一个“通用特征池”。这里的特征只关注“基因本身的语义信息”,而剥离了“这个样本特有的空间分布模式”。这就好比,SASA负责告诉你“苹果”和“橙子”各自是什么(语义),但先不告诉你它们在盘子里具体怎么摆(几何)。摆盘子的通用规则,交给下一个模块去学。
经过这一系列操作,SASA模块产出了一个“通用特征池”。这里的特征只关注“基因本身的语义信息”,而剥离了“这个样本特有的空间分布模式”。这就好比,SASA负责告诉你“苹果”和“橙子”各自是什么(语义),但先不告诉你它们在盘子里具体怎么摆(几何)。摆盘子的通用规则,交给下一个模块去学。物理为先:从无界回归到单纯形比率预测
SRast的第二个核心创新,是重新定义了超分辨率任务本身。它不再预测每个小格子的绝对表达值,而是预测一个分配比率。想象一个大格子总共有100个“苹果基因”分子。超分辨率的本质是:这100个分子,是如何分布到它下属的4个(或9个)小格子里的?是[25, 25, 25, 25]平均分,还是[60, 20, 15, 5]集中在某处?SRast的物理约束流匹配模块,要学的就是这个分配比率向量。这个向量天然位于一个叫“单纯形”的空间里(所有分量非负,且和为1)。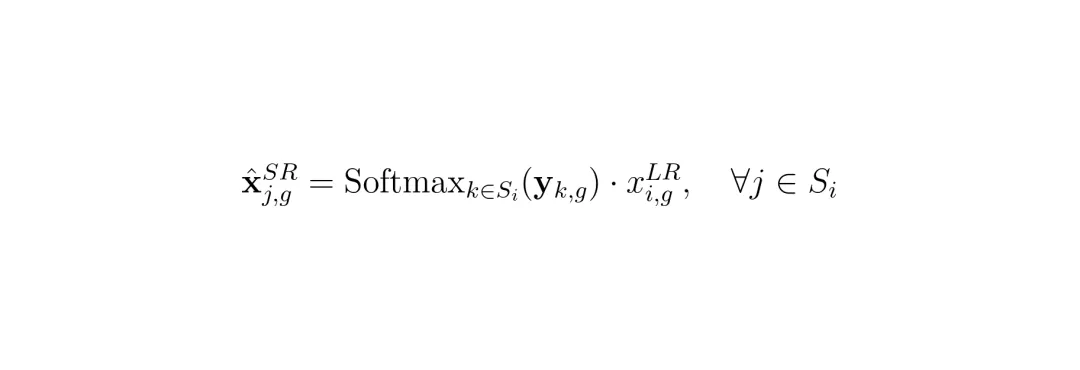 (公式:最终的预测值 = Softmax(预测的对数比) × 低分辨率总表达量。这确保了加和必然等于原值,物理守恒自动满足。)1. 物理一致性成为内置属性:只要预测的是比率,最后乘以总质量,守恒律自动满足,从根本上杜绝了“幻觉”数据。2. 简化了学习目标:模型不再需要学习绝对数值的尺度,只需要学习相对的空间分布模式。不同基因、不同样本的表达量可能相差数个数量级,但它们的空间分布比率可能遵循更通用的几何规律。当然,直接在有边界(0到1)且稀疏(很多比率接近0)的单纯形上做生成模型很困难。SRast巧妙地使用了一个平滑中心化对数比变换,将单纯形上的比率映射到一个无界的、更稳定的欧几里得空间中进行学习,解决了数值计算上的难题。
(公式:最终的预测值 = Softmax(预测的对数比) × 低分辨率总表达量。这确保了加和必然等于原值,物理守恒自动满足。)1. 物理一致性成为内置属性:只要预测的是比率,最后乘以总质量,守恒律自动满足,从根本上杜绝了“幻觉”数据。2. 简化了学习目标:模型不再需要学习绝对数值的尺度,只需要学习相对的空间分布模式。不同基因、不同样本的表达量可能相差数个数量级,但它们的空间分布比率可能遵循更通用的几何规律。当然,直接在有边界(0到1)且稀疏(很多比率接近0)的单纯形上做生成模型很困难。SRast巧妙地使用了一个平滑中心化对数比变换,将单纯形上的比率映射到一个无界的、更稳定的欧几里得空间中进行学习,解决了数值计算上的难题。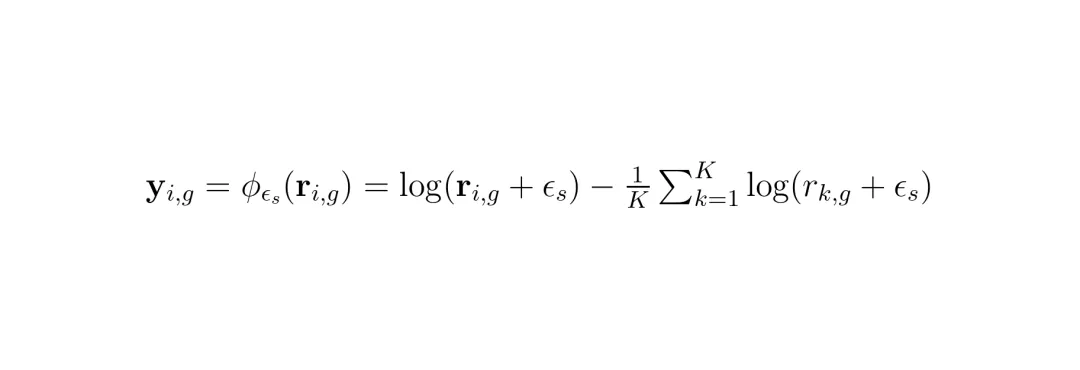 (公式:将比率加上一个极小值取对数,并减去均值,确保数值稳定性。)
(公式:将比率加上一个极小值取对数,并减去均值,确保数值稳定性。)流匹配赋能:学习最优传输的几何变换
现在,任务变成了:在欧几里得空间里,如何从随机噪声生成一个目标比率向量?SRast选择了流匹配这项生成建模技术。你可以把流匹配想象成学习一条从“噪声云”到“目标数据云”的最优传输路径。它不像扩散模型那样需要模拟复杂的随机过程,而是直接学习一个确定的向量场,这个向量场能最平滑、最直接地把噪声点“推”到目标数据点。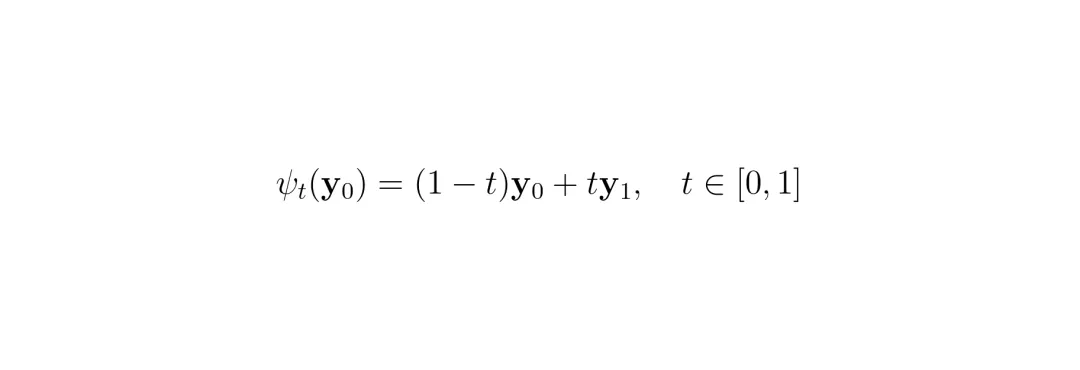 (公式:构建从噪声y₀到目标y₁的线性插值路径 ψ_t。)为了让生成过程可控,PCFM模块将SASA产出的“通用特征”作为条件注入模型。同时,它还注入了高分辨率坐标信息和局部索引信息,告诉模型“现在正在生成哪个小格子”。模型的主干是基于DiT(扩散变换器)架构的,具有强大的全局建模能力。此外,论文还增加了一个空间先验模块,通过一个图神经网络层对高分辨率坐标图进行局部平滑,确保生成的空间分布是合理的(相邻小格子表达量不会突变)。至此,SRast完成了它的设计闭环:SASA提供去除了样本特异性的通用基因语义,PCFM则在物理守恒的硬约束下,学习如何将这些语义依据通用的空间几何规则进行分配。
(公式:构建从噪声y₀到目标y₁的线性插值路径 ψ_t。)为了让生成过程可控,PCFM模块将SASA产出的“通用特征”作为条件注入模型。同时,它还注入了高分辨率坐标信息和局部索引信息,告诉模型“现在正在生成哪个小格子”。模型的主干是基于DiT(扩散变换器)架构的,具有强大的全局建模能力。此外,论文还增加了一个空间先验模块,通过一个图神经网络层对高分辨率坐标图进行局部平滑,确保生成的空间分布是合理的(相邻小格子表达量不会突变)。至此,SRast完成了它的设计闭环:SASA提供去除了样本特异性的通用基因语义,PCFM则在物理守恒的硬约束下,学习如何将这些语义依据通用的空间几何规则进行分配。零样本称王:跨物种、跨平台的强悍泛化
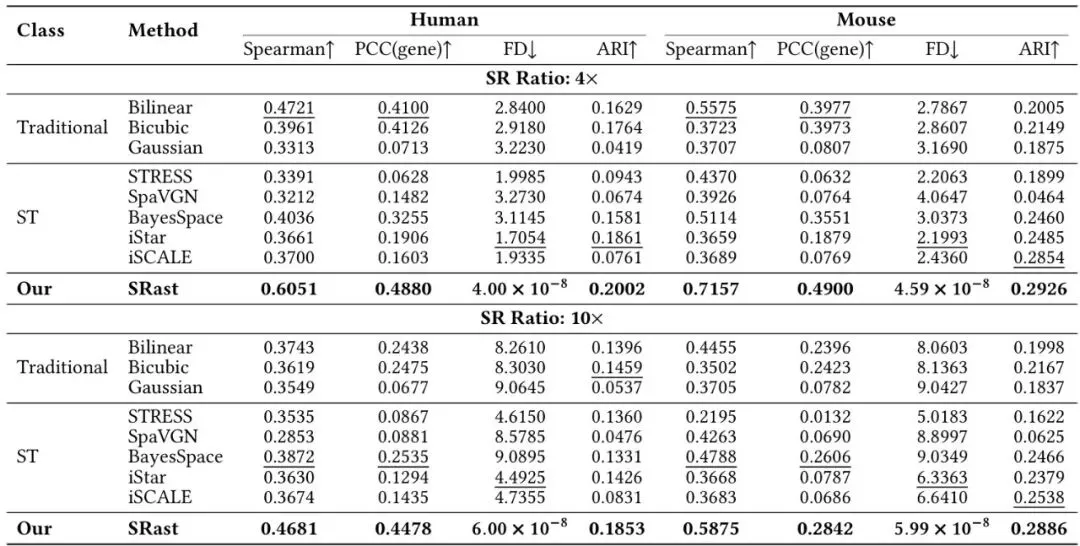
理论很美,实战如何?论文在人类、小鼠等多个物种,以及10x Visium、Slide-seqV2等多种测序平台的数据上进行了零样本测试。零样本的意思是:模型在训练集上从未见过测试组织的任何数据,相当于让模型去“拆一个完全陌生的盲盒”。表1:在人类和小鼠数据集上,跨样本零样本超分辨率性能的量化对比(4倍和10倍上采样)。最佳结果加粗,次佳结果加下划线。↑表示越高越好,↓表示越低越好。Spearman / PCC (gene):衡量预测值与真实值在基因层面的相关性。SRast在绝大多数情况下大幅领先所有基线方法。FD (Fréchet Distance):衡量整体分布的一致性。注意看SRast的结果——6.00 x 10⁻⁸!这几乎为零的值,完美印证了其物理约束的有效性,预测分布与真实分布在整体上几乎重合。而其他方法FD值很大,说明产生了分布偏移。ARI (Adjusted Rand Index):衡量预测的空间区域(如细胞群落)与真实区域的匹配度。SRast也 consistently 表现优秀。在更极端的跨物种(训练用人类/小鼠,测试用斑马鱼)和跨平台(训练用10x Visium,测试用Slide-seqV2)实验中,SRast同样展现出断层式的领先优势,其他方法在OOD场景下性能暴跌,而SRast稳如泰山。效率与一致性兼得:SRast的实用价值
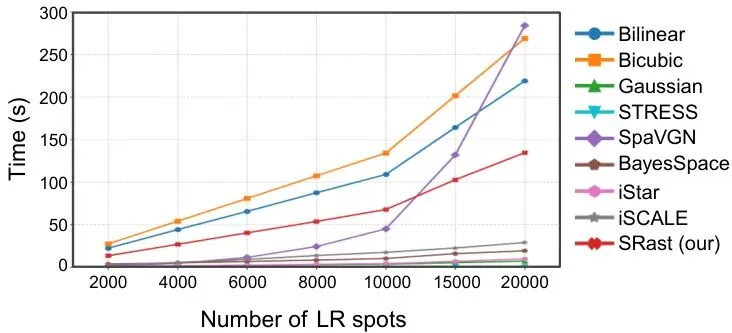
一个强大的模型如果推理速度极慢,实用价值也会大打折扣。论文对比了SRast与基线方法在处理越来越多样本时的运行时间成本。图3:随着推理样本数增加,SRast与基线方法的运行时间成本对比。可以看到,SRast(红色曲线)的推理时间随着样本量增长非常平缓,效率远高于iStar、BayesSpace等方法。这使得它能够处理大规模的数据集,具备了实际应用的可能性。1. 开箱即用:无需针对每个新组织重新训练,一个通用模型解决多种问题,极大降低了使用门槛和计算成本。2. 结果可信:物理守恒约束保证了预测结果在总量上是准确的,避免了误导性的生物学发现。3. 效率可观:推理速度快,能满足实际研究中处理大量数据的需要。龙迷三问
空间转录组和普通转录组有什么区别?普通转录组(Bulk RNA-seq)是把一堆细胞磨碎了测序,得到的是所有细胞的“平均”基因表达。单细胞转录组(scRNA-seq)能测单个细胞,但丢失了细胞在组织中的原始位置信息。空间转录组(ST)则在测量基因表达的同时,保留了每个测量点在组织切片上的精确坐标,是真正能反映“空间异质性”的技术。
论文里总提到的OOD是什么意思?OOD是Out-of-Distribution的缩写,中文可译为“分布外”。指测试数据与模型训练时所看到的数据来自不同的概率分布。在本文中,训练数据来自人类/小鼠的某些组织,测试时用的斑马鱼组织或不同测序平台的数据,就是典型的OOD场景。模型在OOD数据上表现的好坏,直接衡量了其泛化能力的强弱。
流匹配和扩散模型有什么区别?它们都是强大的生成模型。扩散模型通过模拟一个逐步加噪(前向过程)和逐步去噪(反向过程)的随机过程来生成数据。而流匹配则更直接,它学习一个确定的向量场,这个向量场定义了从简单噪声分布到复杂数据分布的最优传输路径。流匹配通常训练更稳定,采样步骤可以更少,效率更高。本文用流匹配来学习基因表达比率的分布,非常合适。
如果你还有哪些想要了解的,欢迎在评论区留言或者讨论~龙哥点评
论文创新性分数:★★★★☆
将物理守恒约束作为硬条件引入生成过程,并采用解耦架构应对生物异质性,思路清晰且有深度,不是简单的模型堆砌。实验合理度:★★★★★
实验设计非常扎实,不仅做了常规对比,还重点突出了零样本跨物种、跨平台这种极具挑战性的OOD泛化测试,结论说服力强。学术研究价值:★★★★★
为空间转录组分析乃至更广泛的生物信息学生成任务提供了一个兼具理论严谨性(物理约束)和实用性(强泛化)的框架范式,启发意义很大。稳定性:★★★★☆
基于物理约束的设计使其输出在总量上具有内在稳定性,避免了荒谬的结果。但对输入数据质量(如低分辨率数据的准确性)仍有一定依赖。适应性以及泛化能力:★★★★★
论文的核心亮点。跨物种、跨平台的零样本性能证明了其卓越的泛化能力,是迈向“通用”模型的关键一步。硬件需求及成本:★★★☆☆
基于Transformer的流匹配模型训练成本不低。但推理效率尚可,且一次训练后可广泛复用,拉平了使用成本。复现难度:★★★☆☆
方法涉及多个复杂模块(GVAE, 流匹配, DiT),复现有一定技术门槛。若能开源代码和预训练模型,将极大降低难度。产品化成熟度:★★★☆☆
作为研究原型已非常出色。要产品化,还需在更多真实世界、有噪声的数据上进行验证,并可能需要对极端情况进行工程化加固。可能的问题:
方法主要依赖已有的大量跨物种数据来学习“通用规则”。对于全新出现的、与训练数据模式迥异的生物结构或疾病状态,其泛化能力边界有待探索。此外,解耦的彻底性也可能影响对某些高度特异的空间模式的捕捉。[1] Xinlei Huang, Weihao Dai, Zijun Qin, et al. Towards Universal Spatial Transcriptomics Super-Resolution: A Generalist Physically Consistent Flow Matching Framework. arXiv:2602.10644v1, 2026. (本论文)[6] BayesSpace 等经典及SOTA空间转录组超分辨率方法,详见论文引用部分。[23] Diffusion Transformer (DiT) 等相关生成模型工作。*本文仅代表个人理解及观点,不构成任何论文审核或者项目落地推荐意见,具体以相关组织评审结果为准。欢迎就论文内容交流探讨,理性发言哦~ 想了解更多原文细节的小伙伴,可以点击左下角的"阅读原文",查看更多原论文细节哦!

想和更多AI医疗、生物信息学的小伙伴交流SRast这样的前沿技术吗?欢迎加入龙哥读论文粉丝群,
扫描下方二维码或者添加龙哥助手微信号加群:kangjinlonghelper。
一定要备注:研究方向+地点+学校/公司+昵称(如 AI医疗+深圳+港中文+龙哥),根据格式备注,可更快被通过且邀请进群。



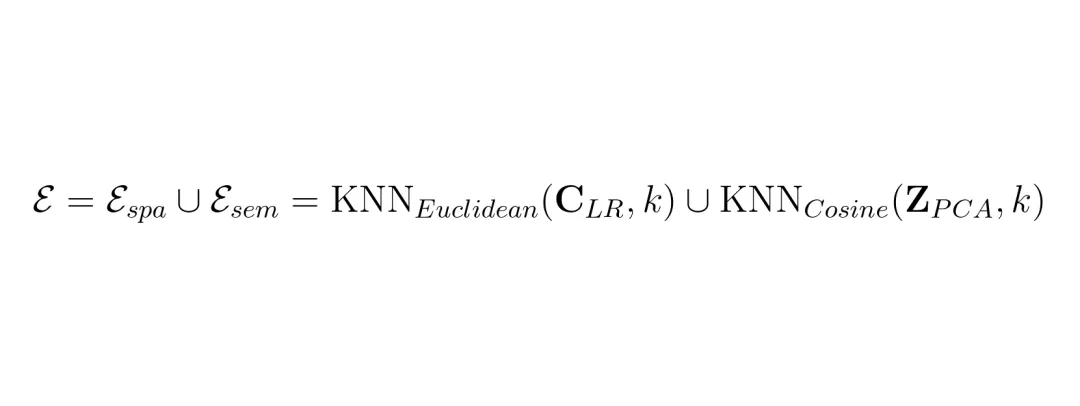



 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
