1、文章亮点
1、贵金属掺杂NiV层状双氢氧化物(LDHs)中,优化了活性位点的电子结构,显著提升了其析氢反应(HER)和析氧反应(OER)的催化性能;
2、提出 “双氯限域” 策略,通过强吸附作用(Ir-Cl)与静电排斥作用(VO43-)的协同作用,克服海水电解中的氯离子腐蚀问题;
3、为经济可行且可持续的海水制氢提供了一条实用路径。
2、全文速览
海水电解是可持续制氢的理想路径,但氯离子诱导的严重腐蚀和寄生氯氧化反应阻碍了其实际应用。本文报道了一种贵金属掺杂的NiV 层状双氢氧化物(LDHs)催化剂,整合了电子调控与双氯限域机制:Ir 掺杂同时构建了强 Ir-Cl 配位作用和动态再生的 VO43-层,形成适应性静电屏障,有效抑制氯离子渗透,使Ir-NiV LDH 的析氧反应选择性接近 100%,在 500 mA・cm-2电流密度下稳定性超2750 小时;Ru 掺杂优化了析氢反应路径,实现 195 mV 的低过电位和超过 2350 小时的耐久性。将两种催化剂配对构建双电极电解槽(Ru-NiV LDH||Ir-NiV LDH),在碱性海水中展现出工业级性能和卓越稳定性。该双氯限域概念为腐蚀性离子环境中的催化剂设计提供了通用框架,可拓展至海水分解以外的其他电化学能量转换过程。
3、背景介绍
1、研究基础:电化学水分解是大规模可持续制氢的核心技术,但现有电解槽依赖高纯度淡水,限制了在缺水地区的部署;海水占地球水资源的 96.5%,是近乎取之不尽的原料,直接利用海水进行电解制氢具有重要意义。
2、核心挑战:海水中高浓度氯离子(~0.5 M)在碱性条件下使热力学电位窗口缩小约 490mV,导致寄生氯氧化反应(ClOR)优先于析氧反应(OER)发生,同时氯离子吸附和腐蚀会加速催化剂降解,如何获得高活性、高稳定性、高选择性的海水电解催化剂是当前研究的核心难题。
3、现有方案不足:已报道的催化剂设计策略(如异质结工程、杂原子掺杂、缺陷工程)中,贵金属掺杂因能调控电子结构和中间体吸附能而展现潜力,但现有斥氯策略多依赖单一机制(物理排斥或化学限制),在工业所需的高电流密度下易因浓度梯度和氯离子渗透而失效。
4、研究动机:针对上述问题,本文设计了 “电子调控 + 双氯限域” 协同的贵金属掺杂 NiV LDH 催化剂,解决海水电解的稳定性和选择性瓶颈。
4、图文解析
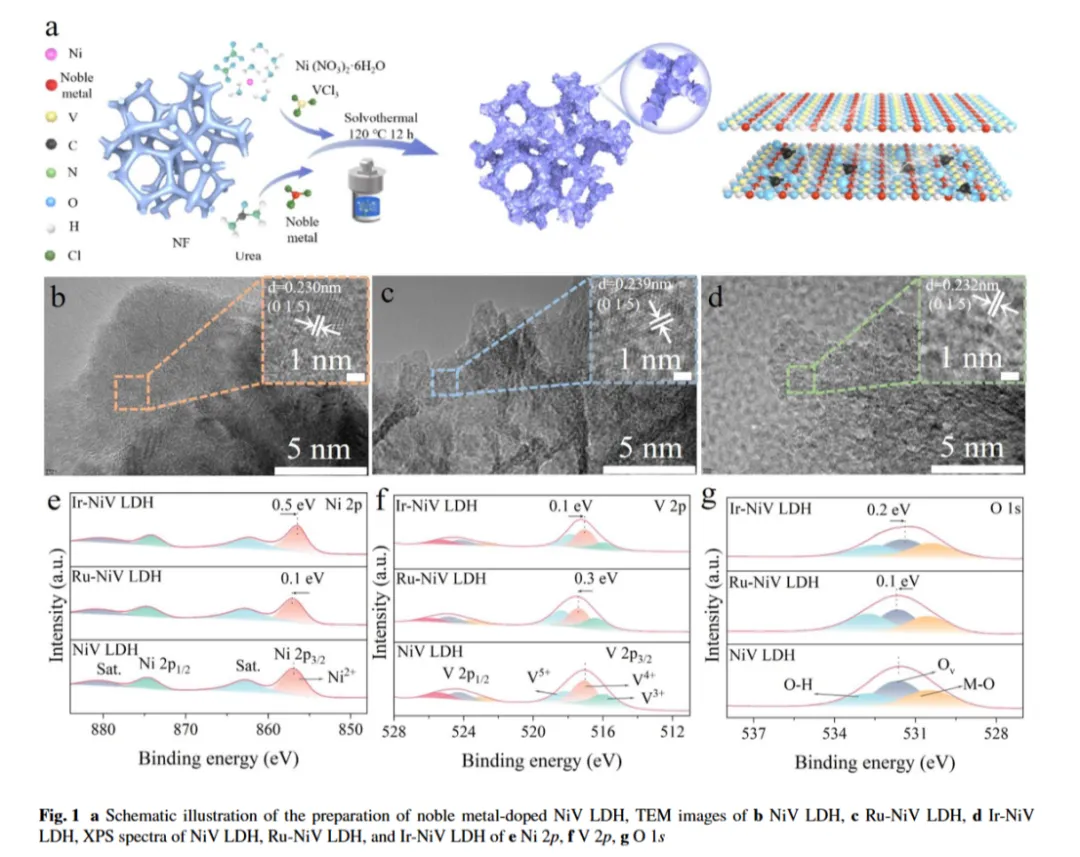
图1a:目标催化电极合成路径及电子调控示意图
图2b-d:高分辨透射电子显微镜(HR-TEM)图像:NiV LDH、Ru-NiV LDH 和 Ir-NiV LDH 的晶格条纹间距分别为 0.230 nm、0.239 nm 和 0.232 nm,均对应于(015)晶面
图1e:高分辨Ni 2p XPS 图谱:856.9 eV 和 874.5 eV 处的峰为Ni2+的特征峰,伴随861.9 eV 和 880.0 eV 处的卫星峰。与未掺杂 NiV LDH 相比,Ru-NiV LDH 和 Ir-NiV LDH 的 Ni 2p 结合能分别向低能端偏移约 1.1 eV 和 0.6 eV,表明贵金属成功掺杂并调控了 NiV LDH 的电子结构,这种电子偏移可能源于贵金属掺杂后表面电子密度的变化
图1f:V 2p 图谱:516.2 eV、517.4 eV 和 519.0 eV 处的三个特征峰分别对应 V3+、V4+和V5+物种,Ru-NiV LDH 和 Ir-NiV LDH 中这些峰的结合能分别偏移 1.2 eV 和 0.2 eV,进一步证明界面存在强烈的电子相互作用
图1g:NiV LDH 的高分辨O1s 图谱:可分解为530.7 eV(金属-氧键,M-O)、531.9 eV(氧空位)和533.5 eV(吸附氧)三个峰;Ru-NiV LDH 和 Ir-NiV LDH 中这些峰分别向低能端偏移 1.1 eV 和 0.7 eV,进一步佐证了界面电子相互作用
结论:上述表征均表明Ru 和 Ir 在掺杂催化剂中均匀分布
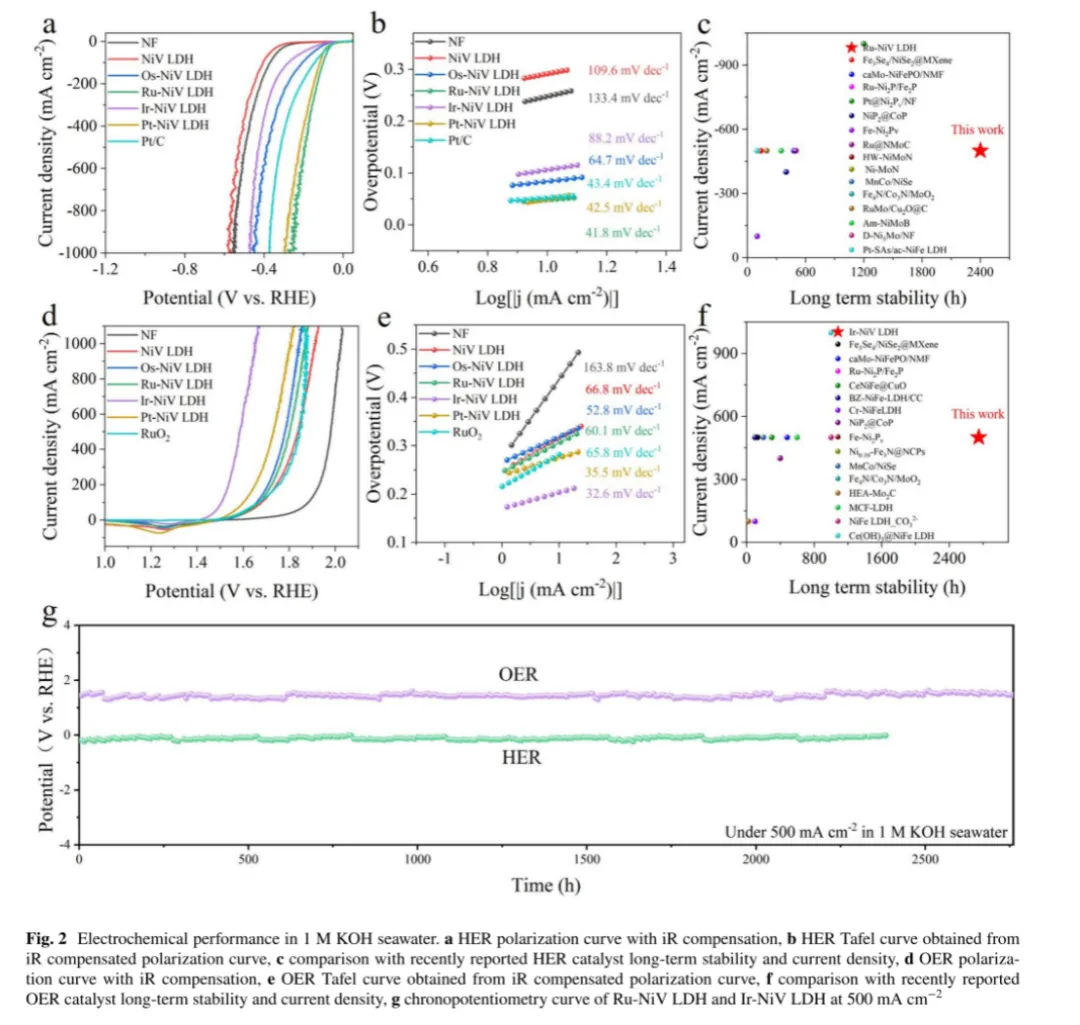
图2a:泡沫镍(NF)、NiV LDH、Os-NiV LDH、Ru-NiV LDH、Ir-NiV LDH、Pt-NiV LDH 及商业Pt/C催化剂的 iR 补偿极化曲线:Ru-NiV LDH 表现出最高的 HER 催化活性:在 500 mA・cm-2的高电流密度下,其过电位仅为195 mV,显著优于商业Pt/C 催化剂(相同电流密度下过电位为 316 mV),过电位降低了 121 mV
图2b:通过iR 补偿极化曲线推导的塔菲尔(Tafel)斜率:Ru-NiV LDH 的 Tafel 斜率为 41.8 mV・dec-1,显著低于未掺杂NiV LDH(109.6 mV・dec-1)和商业Pt/C(43.4 mV・dec-1),表明Ru 掺杂显著加速了 HER 动力学过程
图2c和d:大多数先进HER 电催化剂在超过 500 mA・cm-2的电流密度下难以维持长期稳定性,通常无法在该高电流密度下连续运行超过1000 h,但本工作可在超过500 mA・cm-2的电流密度下持续工作超两千小时,凸显出本工作重要的意义。
图2d:OER催化性能:Ru-NiV LDH 表现出最佳HER催化性能,而 Ir-NiV LDH 在 1 mol/L KOH 海水中展现出卓越的OER活性。测试了 NF、NiV LDH、Os-NiV LDH、Ru-NiV LDH、Ir-NiV LDH、Pt-NiV LDH 及商业 RuO₂的iR补偿OER极化曲线,Ir-NiV LDH 在 10 mA・cm-2电流密度下的过电位仅为202 mV,显著低于商业 RuO2(286 mV);在 500 mA・cm-2的工业级电流密度下,Ir-NiV LDH 的过电位为 357 mV,远低于商业 RuO₂(614 mV)。在OER 极化曲线的反向扫描过程中,1.23 V 处出现一个氧化还原峰,对应 Ni3+向Ni2+的阳极还原反应。考虑到水氧化前催化剂表面结构的动态重构,研究不同催化剂中金属物种的氧化还原电化学行为至关重要,这可能对活性位点的形成产生决定性影响
图2e:塔菲尔斜率:Ir-NiV LDH 的塔菲尔斜率为 32.6 mV・dec-1,低于NiV LDH(66.8 mV・dec-1),表明Ir 掺杂优化了电子结构并加速OER过程
图2g:在500 mA・cm-2的工业级电流密度下,Ru-NiV LDH 在 2350 h 的 HER 稳定性测试中表现出卓越的稳定性,衰减速率仅为 0.072 mV・h-1。测试过程中的电位波动可能源于电解液补充、温度变化或气泡干扰等因素
结论:这些结果表明Ir-NiV LDH 在海水电解中表现出优异的OER活性和长期稳定性,在工业水分解应用中具有巨大潜力
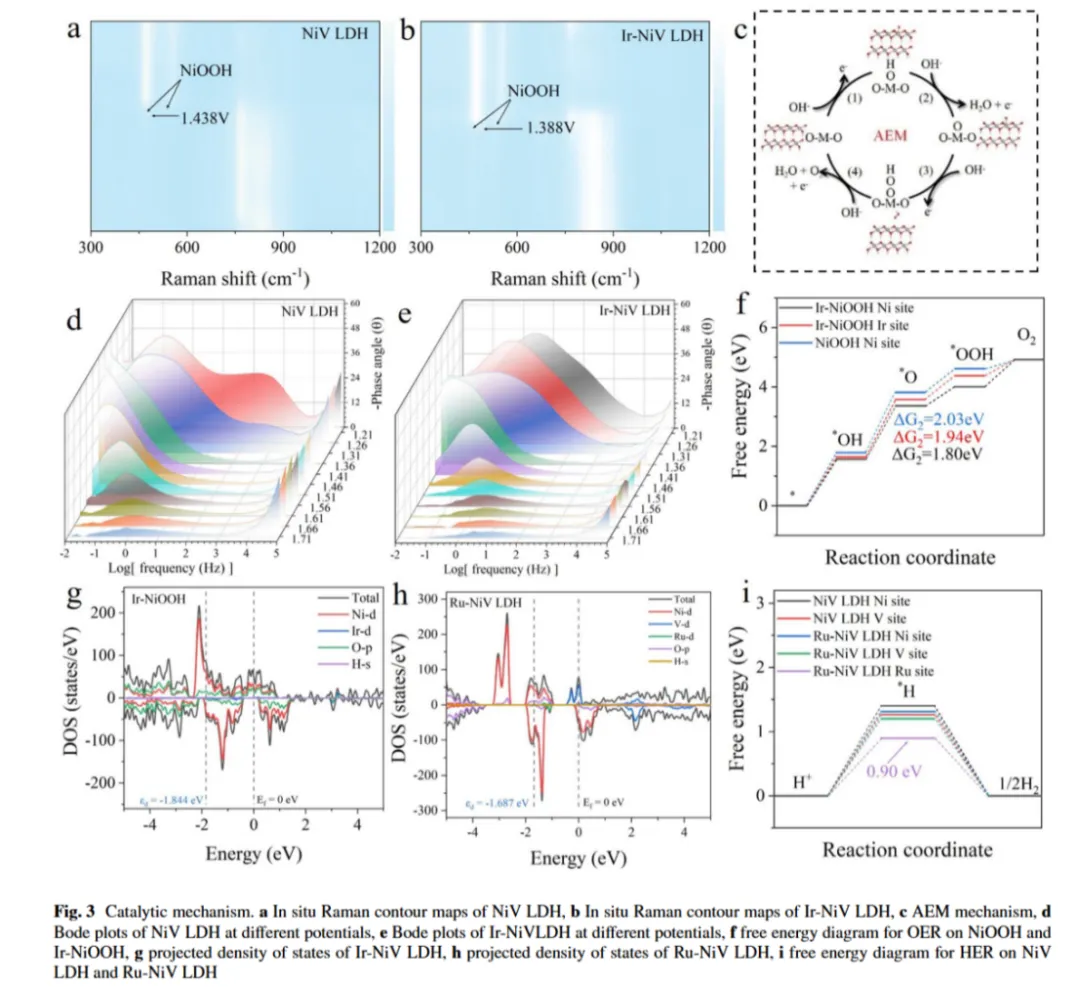
图3a和b:在1 mol/L KOH 电解液中,NiV LDH 在 1.438 V 阳极电位下出现 474 cm-1和554 cm-1的新拉曼峰,这些峰归属于NiOOH相。在 OER 过程中,NiOOH通常被认为是真正的催化活性物种,且NiOOH的早期重构与更优的催化活性相关。相比之下,Ir-NiV LDH 的NiOOH特征峰在更低的 1.388 V 电位下出现,表明 Ir 掺杂促进了Ni (OH)2向NiOOH 的相转变,同时伴随 Ni2+氧化为Ni3+。
图3c:AEM机理:表明Ir-NiV LDH通过优化电子结构和强化中间体吸附能力,显著提升OER性能,为其在海水电解中的应用提供了有价值的理论见解
图3d和e:原位电化学阻抗谱(operando EIS):揭示了OER过程中的界面动力学和电子转移机制,NiV LDH(d图)和Ir-NiV LDH(e图)的波特图均随施加电位增加而发生显著变化:在1.56 V 时,低频区域(10-2-10 Hz)的相角代表 OER 界面,高频区域(10-105Hz)与表面氧化过程相关;低频区域相角降
低,NiV LDH 的相角约为 16°,而 Ir-NiV LDH 约为 6°,表明后者的界面OER速率更快。同时,波特图在低频区域出现一个峰,且随电位增加向低频率偏移,这可归因于 OH * 中间体的吸附;低频区域相角急剧下降的电位对 NiV LDH 为 1.46 V,对 Ir-NiV LDH 为 1.41 V,反映出 Ir-NiV LDH 具有更快的OER速率,与原位拉曼光谱和极化曲线结果一致
图3f:对表面重构后的Ir-NiOOH 的深入分析:Ir-NiOOH 和原始 NiOOH 中的催化活性位点均为暴露的 Ni 中心,中间体(*OH、*O、*OOH)持续吸附。在U=0时,OH向O 转化的活化能垒为 2.03 eV,构成反应的决速步(RDS);而 Ir 掺杂将该能垒降至 1.80 eV(图 3f),表明 Ir 掺入有效调控了 Ni 位点的电子结构,增强了其 OH 吸附能力
图3g:Ir-NiV LDH的投影态密度:选择实际OER 活性物种 NiOOH 和 Ir-NiOOH 作为计算模型,态密度(DOS)分析显示,与 NiOOH 相比,Ir-NiOOH 的态密度在费米能级附近向上偏移,有利于氧位点的电子脱出并降低吸能垒,从而提升OER 活性。
图3h:Ru-NiV LDH的投影态密度:Ru-NiV LDH 的态密度(-1.687 eV)更接近费米能级(0 eV),表明 Ru 掺入增强了电子导电性。这种 d 带中心的调控优化了氢中间体吸附,降低了塔菲尔决速步和氢迁移的能垒。Ru-NiV LDH 的氢吸附吉布斯自由能(GH+)为0.90 eV,显著低于NiV LDH(1.40 eV),表明 Ru-NiV LDH 是最有效的 HER 活性中心
结论:Ru 掺杂诱导的优化电子结构和增强本征活性,凸显了定制化催化剂设计对海水电解中高效稳定 HER 性能的重要性,表明 Ru-NiV LDH 是一种极具潜力的析氢高性能催化剂。此外,通过测量催化剂的表面水接触角评估其亲水性和水捕获能力,结果显示水滴在三种样品表面均能瞬时铺展并完全吸附;值得注意的是,与 NiV LDH 相比,Ir-NiV LDH 和 Ru-NiV LDH 的水滴完全渗透时间显著更短,表明其具有更优异的水捕获能力和增强的亲水性
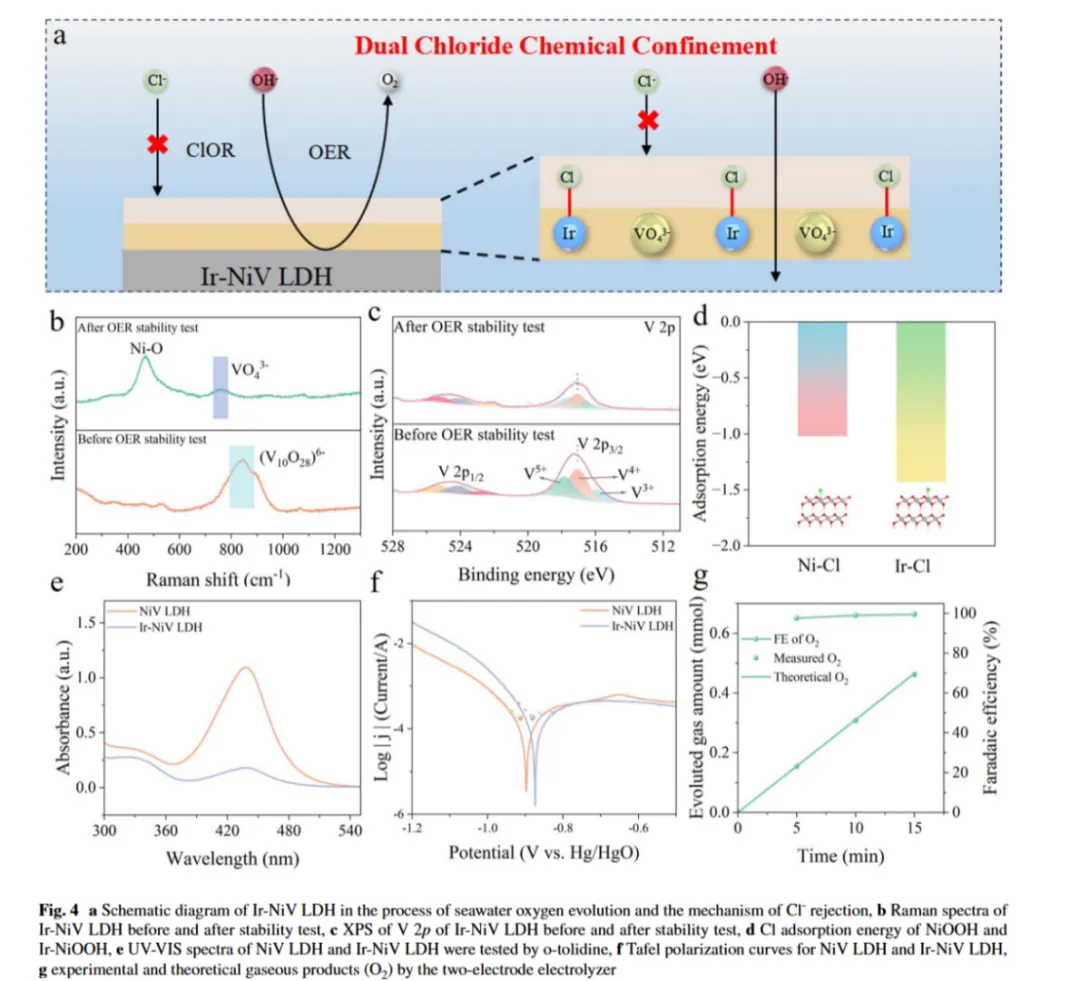
图4a:双斥氯解析图:该结构由原位形成的VO43-层和Ir-Cl 吸附层组成,通过静电排斥协同作用防止 Cl-诱导的腐蚀
图4b:拉曼光谱:证实了Ir-NiV LDH 催化剂中VO43-的存在:对Ir-NiV LDH 催化剂稳定性测试前后的结构变化进行拉曼光谱分析,初始光谱中除两个归属于 Ni-O 振动的特征峰外,843 cm-1处出现一个额外峰,该峰归属于(V10O28)6-物种的对称V-O-V 伸缩振动;稳定性测试后,该峰消失并在760 cm-1处出现新峰,对应VO43-。光谱变化表明V 物种在反应过程中发生溶解,同时 Ir-NiV LDH 从(V10O28)6-转变为VO43-。我们推测,在施加阳极电位下,溶解的V 物种可能重新吸附到电极表面,重构形成保护性 VO43-层,这种动态再生过程有效构建了排斥Cl⁻的静电屏障,从而提升催化剂在含氯电解液中的稳定性
图4c:稳定测试后的XPS光谱:V 2p 图谱显示峰强度显著降低且结合能明显偏移,与 V 物种的溶解一致;稳定性测试后,V3+的相对峰面积显著下降,而V4+和V5+的相对峰面积增加,这种向高氧化态的系统性偏移为(V10O28)6-向VO43-的结构转变相关价态演化提供了关键证据。结合拉曼光谱结果,这些光谱表征共同表明,测试条件下电极表面可能形成了富含V5+的保护层,VO43-可能是其主要成分
图4d:采用DFT 计算研究 Cl 吸附行为,计算结果表明 Ir 位点的氯吸附能力显著增强,吸附能(-1.51 eV)比 Ni 位点(-1.02 eV)更负,这种显著的能量差异证实通过优先化学吸附氯形成了Ir-Cl 保护层
图4e:采用紫外- 可见光谱(UV-vis)、腐蚀电流测试和法拉第效率分析等互补表征技术进一步验证双氯限制机理:在工业电流密度下的系统性 UV-vis 光谱监测显示,与 NiV LDH 对照组相比,Ir-NiV LDH 电解液中活性氯物种的浓度显著更低,表明 Cl⁻氧化得到有效抑制;此外,在含高氯酸盐电解液中进行的计时电流(i-t)测试显示,Ir-NiV LDH 比 NiV LDH 具有更强的结构稳定性,且 ClOR 发生率更低,这些发现进一步证实了 Ir-NiV LDH 固有的氯排斥机理
图4f:通过极化曲线进行的腐蚀评估:评估表明Ir-NiV LDH 比 NiV LDH 具有更高的腐蚀电位和更低的腐蚀电流密度,定量证实其优异的抗氯腐蚀性能。这些结果提供了直接实验证据,表明原位形成的VO43-层和强Ir-Cl 吸附有效保护了电极完整性免受 Cl⁻侵蚀。
图4g:Ir-NiV LDH 电极的法拉第效率:结果显示O₂生成的法拉第效率接近 100%,表明其具有卓越的 OER 选择性
结论:实验结果与理论计算的协同验证了双氯化学限制层的设计有效性,凸显了Ir-NiV LDH 在实际工业海水电解应用中的巨大潜力,解决了海水电解液中稳定性和选择性的双重挑战
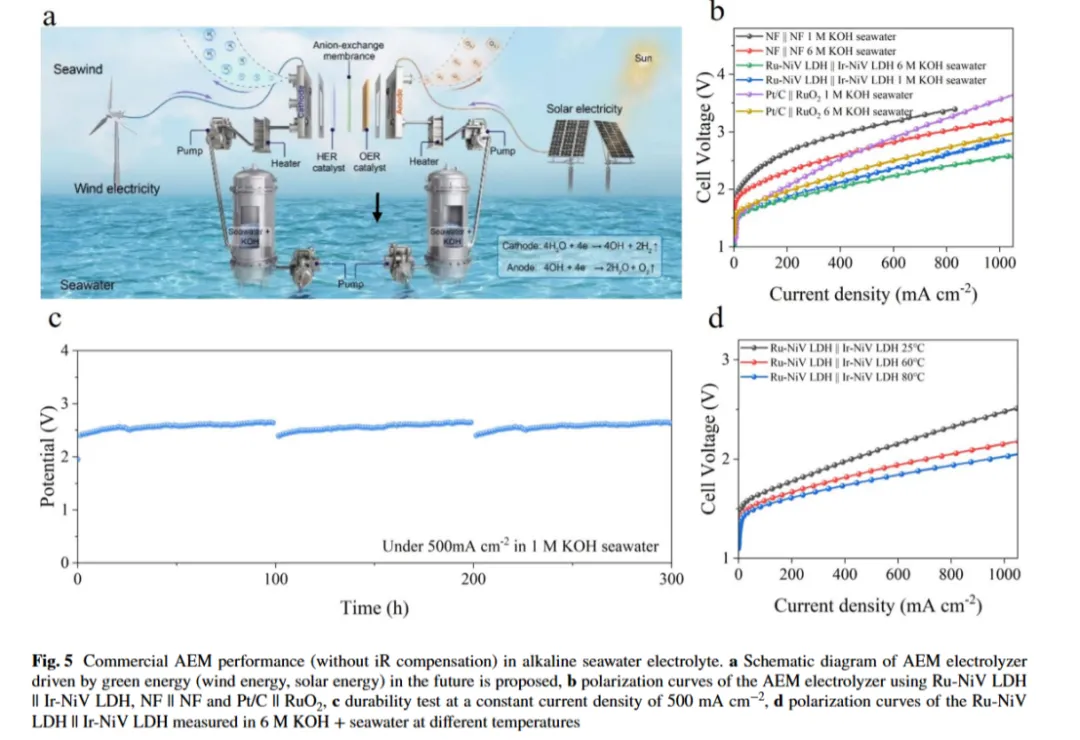
图5a:未来由绿色能源(风能、太阳能)驱动的阴离子交换膜(AEM)电解槽模式图:本研究采用两电极体系(电极面积:1 cm²)和商业电解槽(电极面积:4 cm²)研究了整体海水电解性能。以 Ru-NiV LDH 为阴极、Ir-NiV LDH 为阳极构建两电极体系(Ru-NiV LDH||Ir-NiV LDH),该体系表现出卓越的活性,在仅 2.98 V 的槽电压下实现 500 mA・cm-2的电流密度,性能优于NF||NF 体系(3.60 V)。此外,在 500 mA・cm-2的工业电流密度下对Ru-NiV LDH||Ir-NiV LDH 进行 100 h 海水电解长期稳定性评估,观察到的电位衰减可忽略不计,表明其优异的稳定性。为进一步评估其实际应用潜力,提出了未来由绿色能源(风能、太阳能)驱动的阴离子交换膜(AEM)电解槽模式
图5b:商业电解槽中的Ru-NiV LDH||Ir-NiV LDH 的极化曲线(无 iR 补偿):在500 mA・cm-2电流密度下,Ru-NiV LDH||Ir-NiV LDH 电极在 1 mol/L KOH 海水和 6 mol/L KOH 海水中的整体水分解电压分别为 2.24 V 和 2.14 V,显著低于 NF 电极(3.07 V 和 2.71 V)和 Pt/C||RuO₂参比电极(2.72 V 和 2.38 V),表明其卓越的催化活性
图5c:采用Ru-NiV LDH||Ir-NiV LDH 的商业电解槽在 500 mA・cm-2电流密度下稳定运行超过300 h(图 5c),海水电解过程中未观察到明显故障,表明其潜在的商业价值
图5d:模拟工业条件(6 mol/L KOH + 海水)和不同温度下Ru-NiV LDH 和 Ir-NiV LDH 的极化曲线:结果表明催化剂在80 °C 时活性显著增强,在仅 1.79 V 的槽电压下实现 500 mA・cm-2的电流密度。
结论:Ru-NiV LDH||Ir-NiV LDH 催化剂不仅表现出优异的催化活性和稳定性,还具有显著的经济优势,为其在工业海水电解中的广泛应用提供了有力支持
5、总结与展望
1、总结:合成策略:开发了一步法合成贵金属掺杂 NiV LDH 催化剂的技术,通过精准电子调控和双化学限制机制(强 Ir-Cl 吸附 + 原位生成保护性 VO43-层),实现了腐蚀性海水环境中的高活性和高耐久性;
性能突破:Ru-NiV LDH(析氢)和 Ir-NiV LDH(析氧)在 500 mA・cm-2 工业级电流密度下,过电位分别低至195 mV 和 357 mV,稳定性分别超 2350 小时和 2750 小时;应用潜力:将两种催化剂集成到商业电解槽中,可在更低操作电压下实现高效制氢,展现出显著的工业应用前景。
2、展望:科学价值:深化了对复杂离子环境中催化剂行为的理解,为腐蚀性体系下的催化剂设计提供了通用理论框架;应用拓展:该技术不仅适用于海水制氢,还可推广至其他电化学能量转换过程;产业意义:为规模化、低成本的绿色海水制氢奠定了可行基础,助力可持续氢能产业发展
6、文章链接
《Dual Chloride Confinement in Noble Metal‐Doped NiV LDH Catalysts Enables Stable Industrial‑Level Seawater Electrolysis》
https://doi.org/10.1007/s40820-026-02067-1


如需转载或投稿请联系我们:energy_catalysis@163.com