作者
王莉(天津大学)
吕伟(清华大学深圳国际研究生院)
杨全红(天津大学)
引言
锂硫电池因其高理论能量密度被视为下一代储能系统的有力候选,但其商业化进程受限于硫转化反应的动力学瓶颈和多硫化物的穿梭效应。在多电子转移反应中,催化剂对各类中间体的吸附能往往存在本征线性标度关系,这种约束严重限制了通过调控单一中间体吸附强度来优化催化活性的可能性。现有研究多聚焦于单一电子结构的调控,但对于涉及复杂反应网络和多种中间体(Li2Sn,2≤n≤8)的硫电催化体系,仅调节关键中间体的单一电子结构不足以打破标度关系。因此,建立能够定量反映决速步能量差异并与催化剂电子结构相关联的描述符,对于通过路径级机理调控打破标度关系至关重要。
核心发现
本研究提出了一种基于过渡金属硫化物中金属d带中心与表面硫p带中心匹配程度的预测性描述符——d/p带匹配比。研究发现,该带匹配比与硫还原反应(SRR)和硫析出反应(SER)的过电位呈线性相关,实现了对过渡金属硫化物催化剂活性的定量排序。通过调控带匹配比可优化液-固转化关键步骤的动力学势垒,理论筛选出的NbS2具有99.2%的带匹配比,其双功能过电位总和仅为0.70V,显著优于WS2(2.85V,带匹配比46.0%)。基于NbS2的锂硫电池在高硫载量(9.89mg/cm2)下实现了10.98mAh/cm2的初始面容量,100圈循环后保持率达90.29%,1000圈循环衰减率仅为0.086%/圈。
图文解读
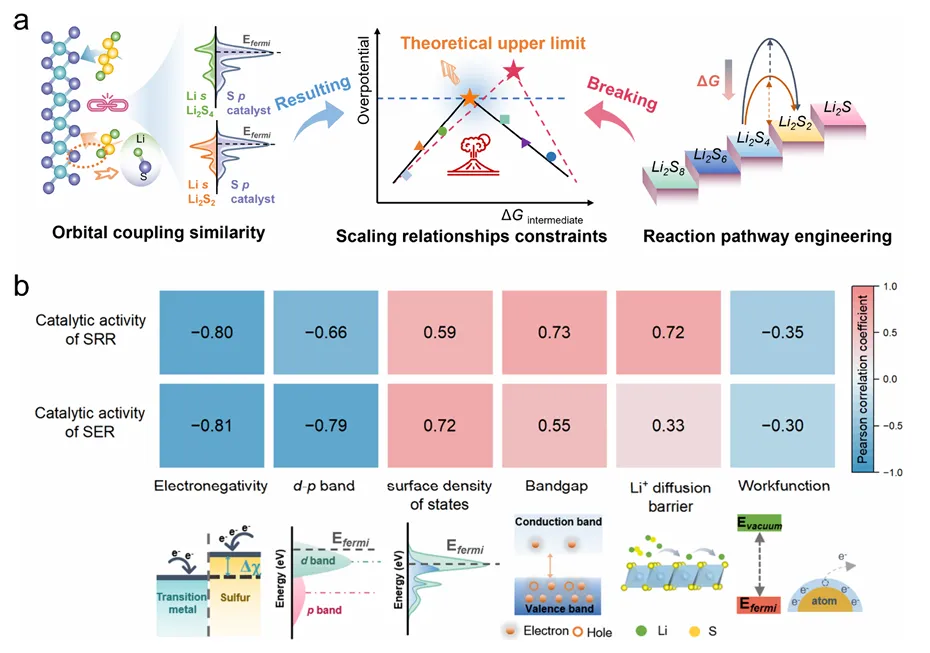
图1:标度关系与路径调控示意图
图1展示了轨道耦合衍生的多硫化物吸附能线性相关对活性优化的限制机制。图中揭示了当催化剂与特定中间体存在强d-p或s-p轨道耦合时,通常对另一中间体表现出相当的吸附强度,从而产生本征线性相关。这种标度关系简化了催化剂筛选,但也限制了通过调节单一中间体结合强度来优化活性的可能性。研究提出通过路径工程聚焦决速步来打破标度关系,这需要调控多种电子结构包括能带结构、原子电负性、功函数、离子电导率和表面态密度等。Pearson相关性分析展示了催化活性与多种电子描述符的相关性,为建立路径依赖描述符提供了理论基础。该分析表明,建立与决速步能量差异相关联的描述符是打破标度关系的关键策略,为锂硫电池电催化剂的理性设计指明了方向。
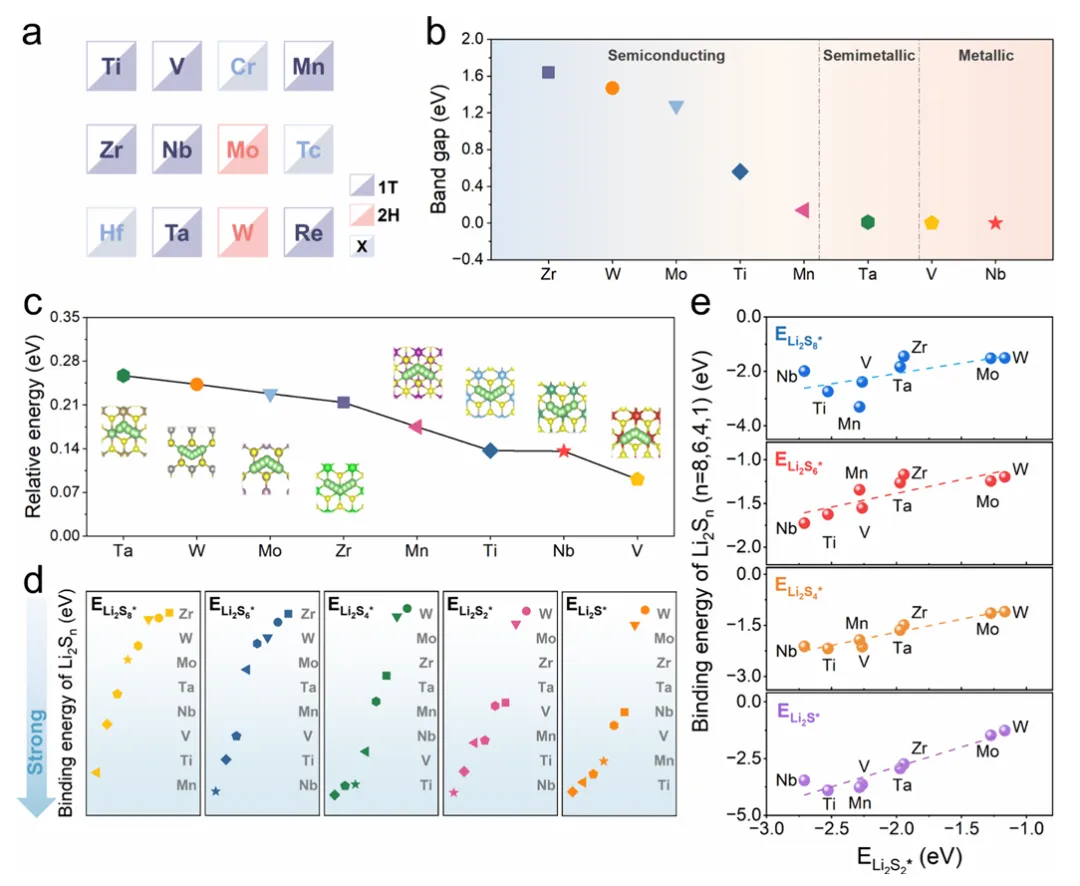
图2:过渡金属硫化物的电子结构与吸附特性
图2系统研究了从IVB到VIIB族的层状过渡金属硫化物的电子结构、锂离子传输特性和吸附能力。研究选取了TiS2、VS2、MnS2、ZrS2、NbS2、MoS2、TaS2和WS2作为模型催化剂,排除了HfS2(带隙过大)、TcS2(放射性)和CrS2(吸附过强且有毒)。能带结构计算显示TMSs的价带顶和导带底之间存在较小的带隙(0-1.64eV),其中NbS2和VS2带隙为0eV,展现出优异的电导率。CI-NEB方法计算的锂离子迁移能垒最大不超过0.26eV,表明所有TMSs均具有优异的锂离子电导率。多硫化物吸附能计算揭示了Li2S8、Li2S6、Li2S4和Li2S的吸附能与Li2S2之间存在线性相关,相关系数达0.97,这种标度关系凸显了多硫化物在TMS表面吸附行为的本征约束,表明仅调节单一中间体吸附强度无法有效调控催化活性。
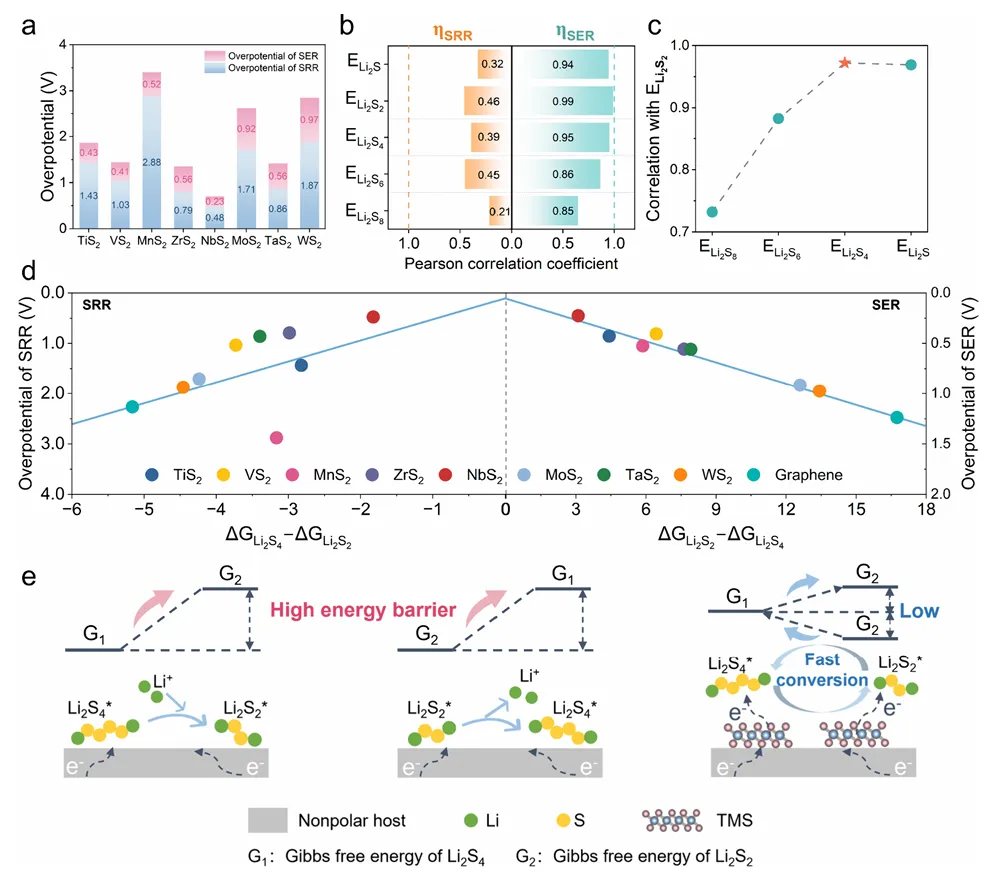
图3:多硫化物吸附与催化活性的关联分析
图3建立了多硫化物吸附能与催化活性之间的关联,提出了通过转化路径调控打破标度关系的策略。理论过电位计算显示,与碳基材料较高的SRR过电位(2.27V)和SER过电位(1.24V)相比,TMS体系的过电位显著降低。其中NbS2在SRR(0.48V)和SER(0.23V)中均具有最低过电位,表明其优异的催化活性。具体而言,NbS2降低了Li2S4到Li2S2的液-固转化能垒,并将SRR的决速步转移到Li2S2到Li2S的固-固转化步骤。Pearson相关性分析表明,单一多硫化物的结合能与SRR过电位的相关性均低于0.5,而与SER过电位的相关性较高(Li2S2的R=0.99)。研究将反应过电位与液-固转化(Li2S4→Li2S2)和固-液转化(Li2S2→Li2S4)的吉布斯自由能差相关联,成功建立了线性关系,其中NbS2位于最优位置。这种路径依赖描述符有效耦合了硫还原和氧化过程,为打破标度关系提供了理论指导。
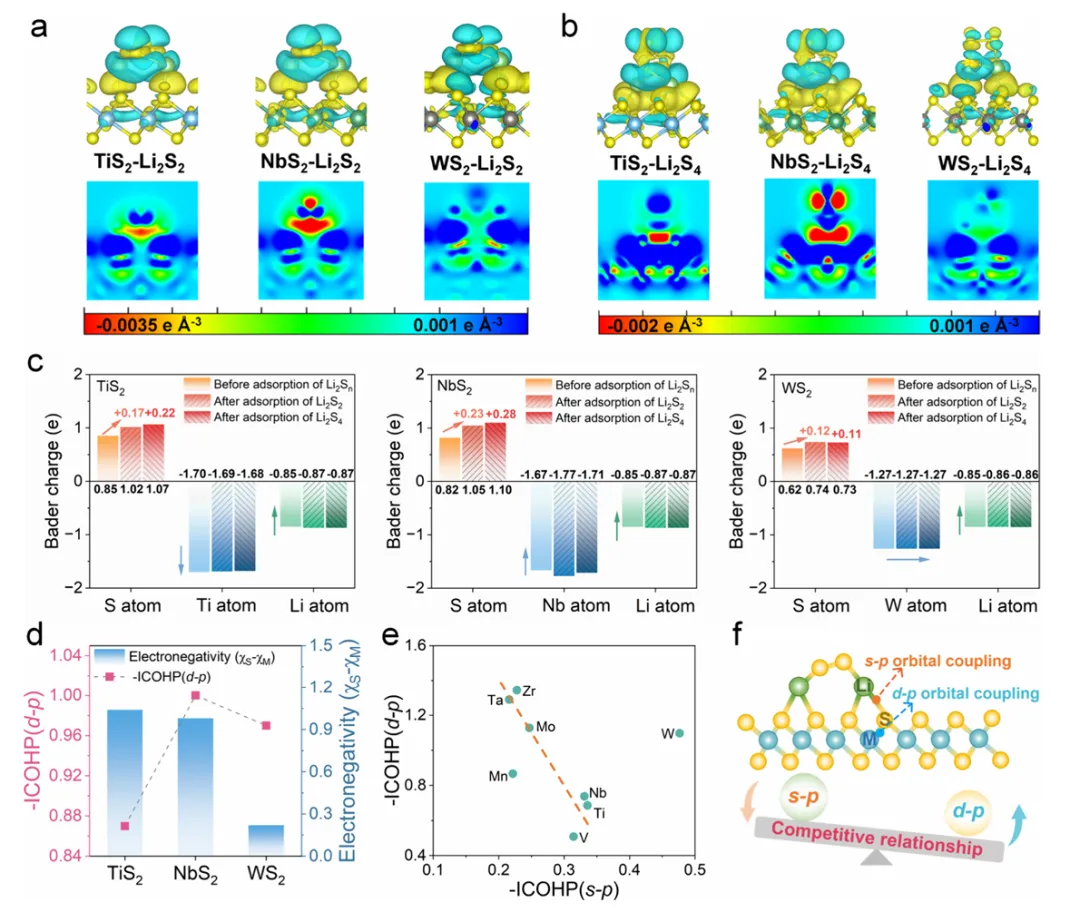
图4:催化活性的电子结构起源
图4通过差分电荷和二维电荷密度图揭示了TMS催化活性的电子结构起源。分析显示Li原子周围的蓝色区域表示电子损失,而TMS中S原子附近的黄色区域表示电子获得。Li2S2和Li2S4内部较少的黄蓝区域表明其内部发生了电荷重分布。三种代表性TMS(Ti、Nb、W)的二维电荷密度图进一步表明,NbS2与Li2S2或Li2S4之间发生最大的电子转移,其次是TiS2,WS2最少,表明NbS2与多硫化物具有最强的电子相互作用,有利于增强反应动力学。Bader电荷分析定量评估了电荷转移:TMS表面S原子获得的电子数分别为TiS2(0.85e)、NbS2(0.82e)和WS2(0.62e)。多硫化物吸附后,Nb原子向表面S原子提供的电子数显著增加(从1.67e增至1.71e),而Ti原子提供的电子减少(从1.70e降至1.68e),W原子基本保持不变(1.27e)。这种差异化的电子转移行为解释了NbS2优异催化活性的来源。
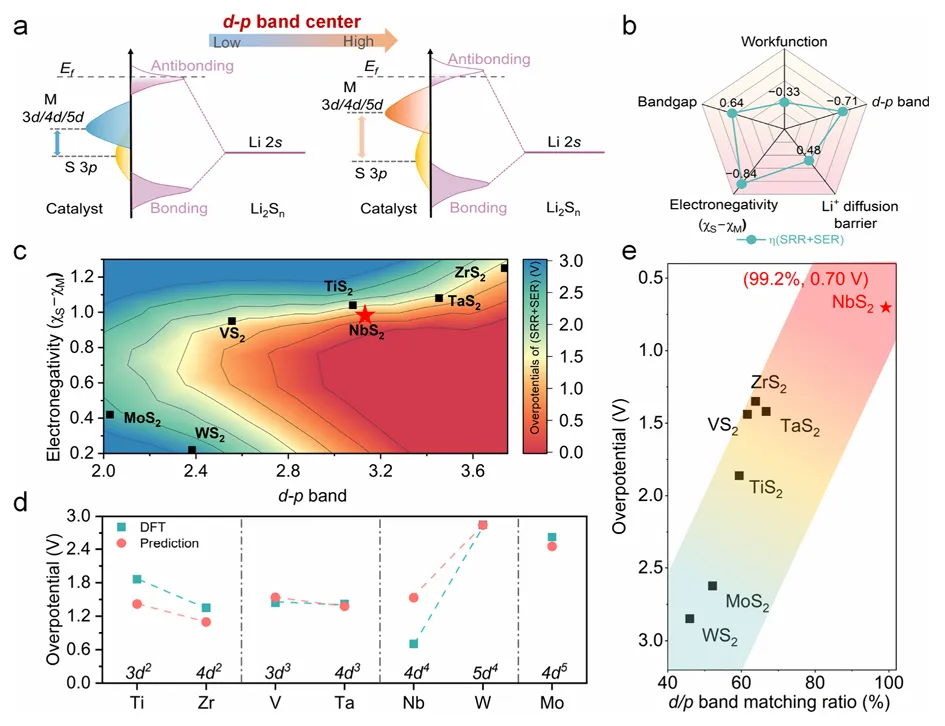
图5:d/p带匹配描述符的建立与验证
图5建立了d/p带匹配比作为硫电催化的预测性描述符。研究通过分析金属d带中心与表面硫p带中心的分离程度,定义了带匹配比来量化d-p轨道对齐程度。COHP分析揭示了M-S键强度与电负性差的关系,表明适中的d-p带中心分离可增强Li-S键合并促进界面电荷转移,而过强或过弱的耦合分别会抑制电子转移或毒化催化位点。带匹配比与SRR和SER过电位之间建立了线性关系,理论预测与实验结果高度吻合。NbS2以99.2%的带匹配比实现了最低的双功能过电位(0.70V),而WS2的带匹配比为46.0%,过电位高达2.85V,两者相差4倍。该描述符成功打破了传统火山型活性限制,为硫电催化剂的理性设计提供了统一原则。优化带匹配可增强表面硫位点的电子密度并促进界面电荷转移,从而降低硫转化的动力学势垒。
总结
本研究建立了d/p带匹配比作为硫电催化的预测性描述符,成功打破了多硫化物吸附能之间的线性标度关系。通过调控带匹配比可优化硫转化反应的动力学势垒,理论筛选并实验验证了NbS2作为最优催化剂,实现了低过电位、高面容量和优异循环稳定性。该工作为金属-硫电池电催化剂的理性设计提供了突破传统火山型限制的新范式。
原文链接
https://doi.org/10.1021/jacs.6c02040


 10个月宝宝每天需要喝多少奶粉?
10个月宝宝每天需要喝多少奶粉?
